

Clinical spectrum of primary hyperoxaluria type 1: Experience of a tertiary center
- PMID: 28161266
- PMCID: PMC5921832
- DOI: 10.1016/j.nephro.2016.08.002
Clinical spectrum of primary hyperoxaluria type 1: Experience of a tertiary center
Abstract
Background and aim: Primary hyperoxalurias are rare inborn errors of metabolism resulting in increased endogenous production of oxalate that leads to excessive urinary oxalate excretion. Diagnosis of primary hyperoxaluria type 1 (PH1) is a challenging issue and depends on diverse diagnostic tools including biochemical analysis of urine, stone analysis, renal biopsy, genetic studies and in some cases liver biopsy for enzyme assay. We characterized the clinical presentation as well as renal and extrarenal phenotypes in PH1 patients.
Methods: This descriptive cohort study included patients with presumable PH1 presenting with nephrolithiasis and/or nephrocalcinosis (NC). Precise clinical characterization of renal phenotype as well as systemic involvement is reported. AGXT mutational analysis was performed to confirm the diagnosis of PH1.
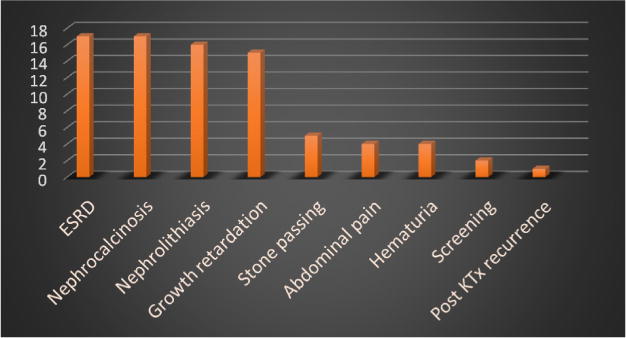
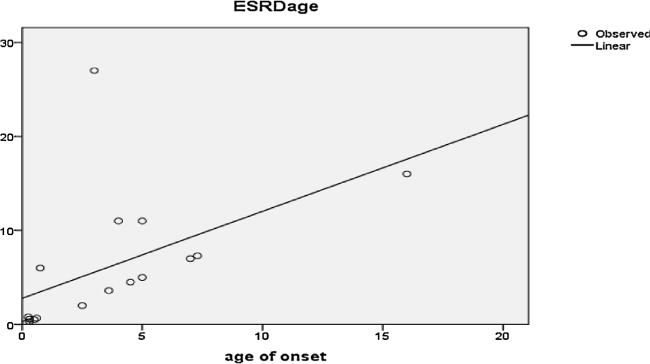
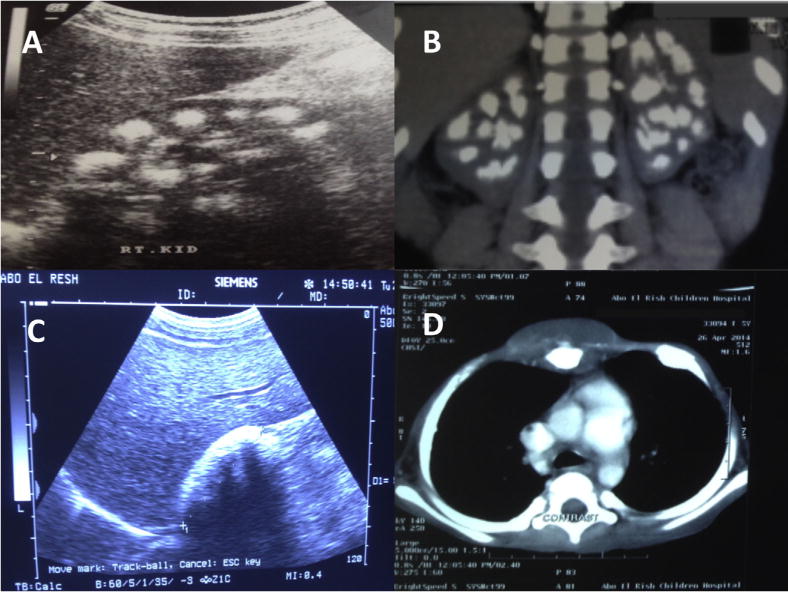
Results: The study cohort included 26 patients with presumable PH1 with male to female ratio of 1.4:1. The median age at time of diagnosis was 6 years, nevertheless the median age at initial symptoms was 3 years. Thirteen patients (50%) were diagnosed before the age of 5 years. Two patients had no symptoms and were diagnosed while screening siblings of index patients. Seventeen patients (65.4%) had reached end-stage renal disease (ESRD): 6/17 (35.3%) during infancy, 4/17 (23.5%) in early childhood and 7/17 (41.29%) in late childhood. Two patients (7.7%) had clinically manifest extra renal (retina, heart, bone, soft tissue) involvement. Mutational analysis of AGXT gene confirmed the diagnosis of PH1 in 15 out of 19 patients (79%) where analysis had been performed. Fifty percent of patients with maintained renal functions had projected 10 years renal survival.
Conclusion: PH1 is a heterogeneous disease with wide spectrum of clinical, imaging and functional presentation. More than two-thirds of patients presented prior to the age of 5 years; half of them with the stormy course of infantile PH1. ESRD was the commonest presenting manifestation in two-thirds of our cohort.
Keywords: End-stage renal disease; Nephrocalcinosis; Nephrolithiasis; Oxalosis; Post-transplantation recurrence; Primary hyperoxaluria type 1.
Copyright © 2016 Association Société de néphrologie. Published by Elsevier Masson SAS. All rights reserved.
Conflict of interest statement
The authors declare that they have no competing interest.
Figures
References
-
- Danpure CJ, Jennings PR. Peroxisomal alanine: glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Lett. 1986;201:20–4. - PubMed
-
- Purdue PE, Lumb MJ, Fox M, Griffo G, Hamon-Benais C, Povey S, et al. Characterization and chromosomal mapping of a genomic clone encoding human alanine: glyoxylate aminotransferase. Genomics. 1999;10:34–42. - PubMed
-
- Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. The gene encoding hydroxypyruvate reductase (GRHPR) is mutated in patients with primary hyperoxaluria type II. Hum Mol Gen. 2009;8:2063–9. - PubMed
-
- Belostotsky R, Pitt JJ, Frishberg Y. Primary hyperoxaluria type III–a model for studying perturbations in glyoxylate metabolism. J Mol Med. 2012;90:1497–504. - PubMed
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
