

Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras
- PMID: 28162770
- PMCID: PMC5445660
- DOI: 10.1016/j.cell.2017.01.013
Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras
Abstract
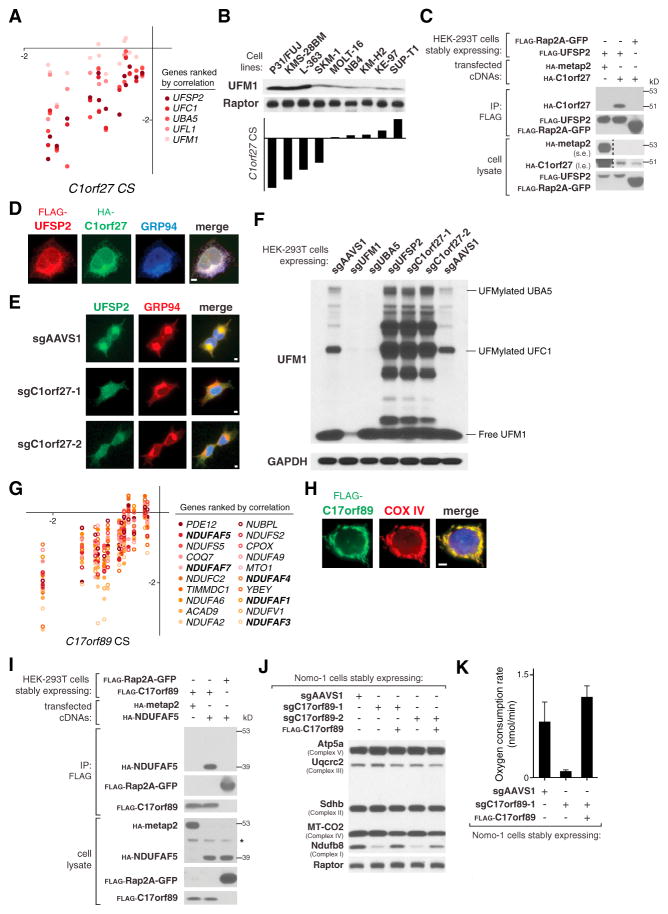
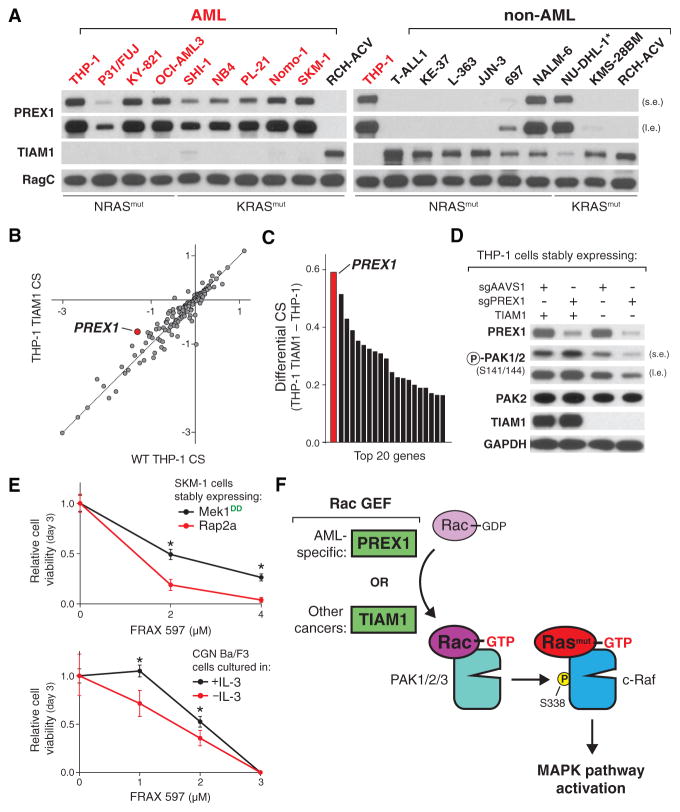
The genetic dependencies of human cancers widely vary. Here, we catalog this heterogeneity and use it to identify functional gene interactions and genotype-dependent liabilities in cancer. By using genome-wide CRISPR-based screens, we generate a gene essentiality dataset across 14 human acute myeloid leukemia (AML) cell lines. Sets of genes with correlated patterns of essentiality across the lines reveal new gene relationships, the essential substrates of enzymes, and the molecular functions of uncharacterized proteins. Comparisons of differentially essential genes between Ras-dependent and -independent lines uncover synthetic lethal partners of oncogenic Ras. Screens in both human AML and engineered mouse pro-B cells converge on a surprisingly small number of genes in the Ras processing and MAPK pathways and pinpoint PREX1 as an AML-specific activator of MAPK signaling. Our findings suggest general strategies for defining mammalian gene networks and synthetic lethal interactions by exploiting the natural genetic and epigenetic diversity of human cancer cells.
Keywords: AML; CRISPR; RAS; gene networks; genetic screens; synthetic lethality.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures





References
-
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang C-Z, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. Genomic copy number dictates a gene-independent cell response to CRISPR-Cas9 targeting. Cancer Disc. 2016 http://dx.doi.org/10.1158/2159-8290.CD-16-0154. - DOI - PMC - PubMed
-
- Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell. 2011;145:30–38. - PubMed
-
- Bassi DE, Fu J, Lopez de Cicco R, Klein-Szanto AJP. Pro-protein convertases: “master switches” in the regulation of tumor growth and progression. Mol Carcinog. 2005;44:151–161. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
