

The dilemma of diagnostic testing for Prader-Willi syndrome
- PMID: 28164030
- PMCID: PMC5253259
- DOI: 10.21037/tp.2016.07.04
The dilemma of diagnostic testing for Prader-Willi syndrome
Abstract
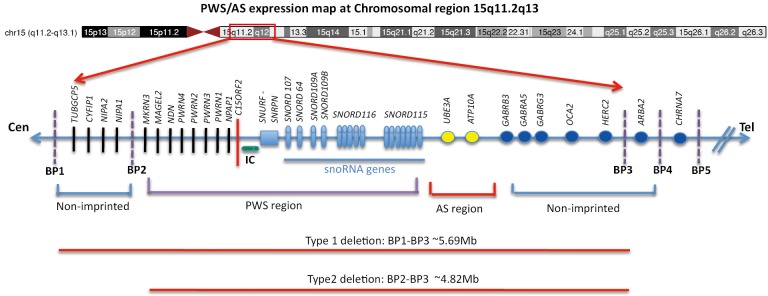
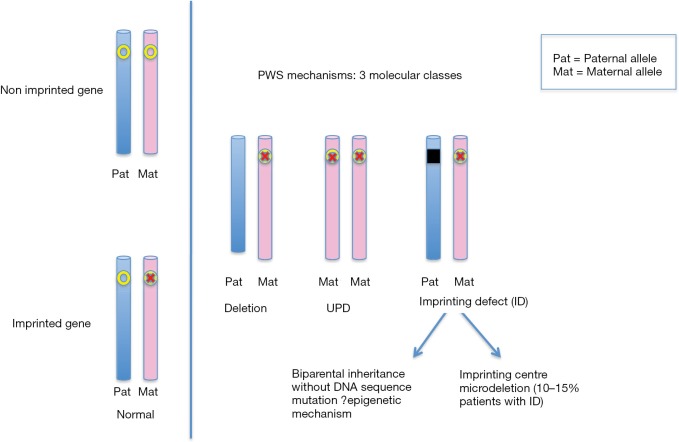
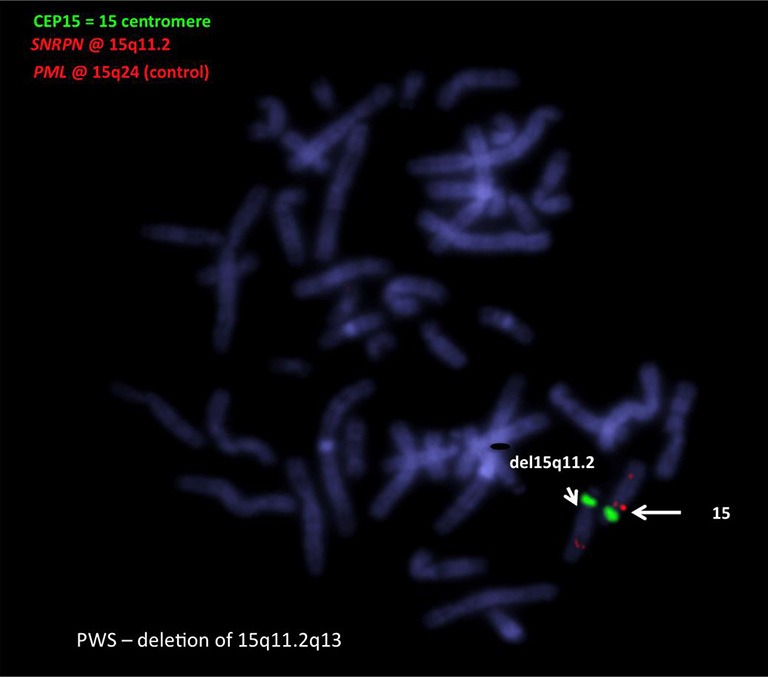
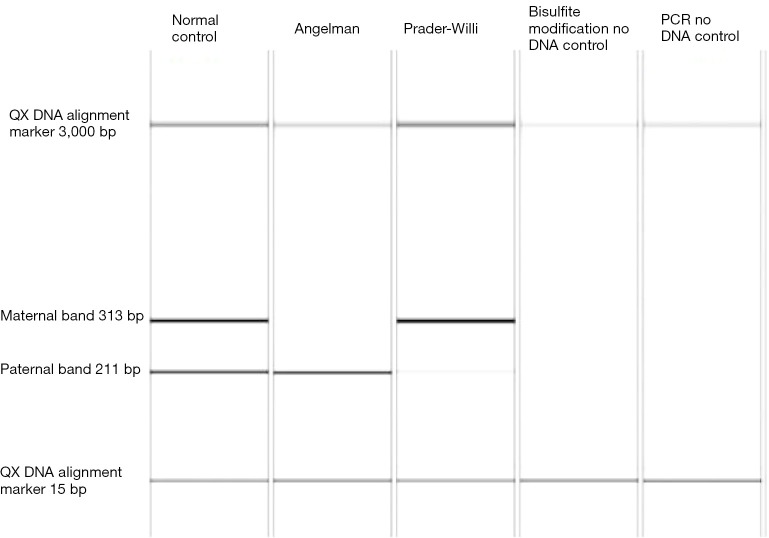
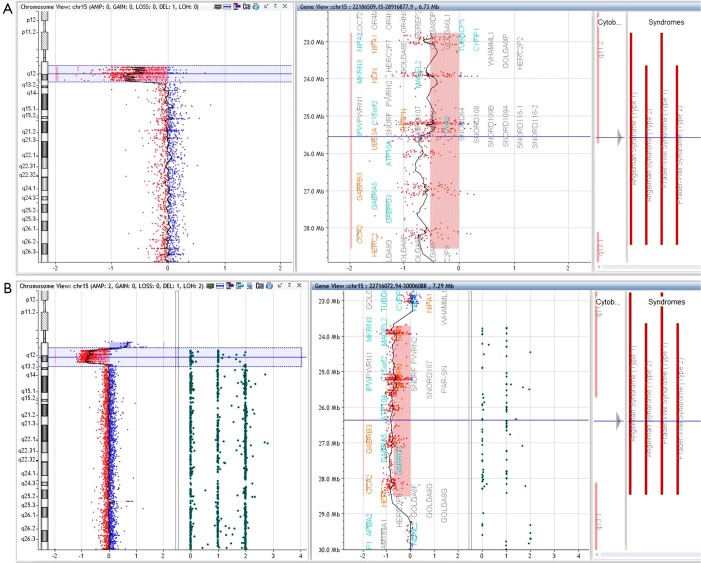
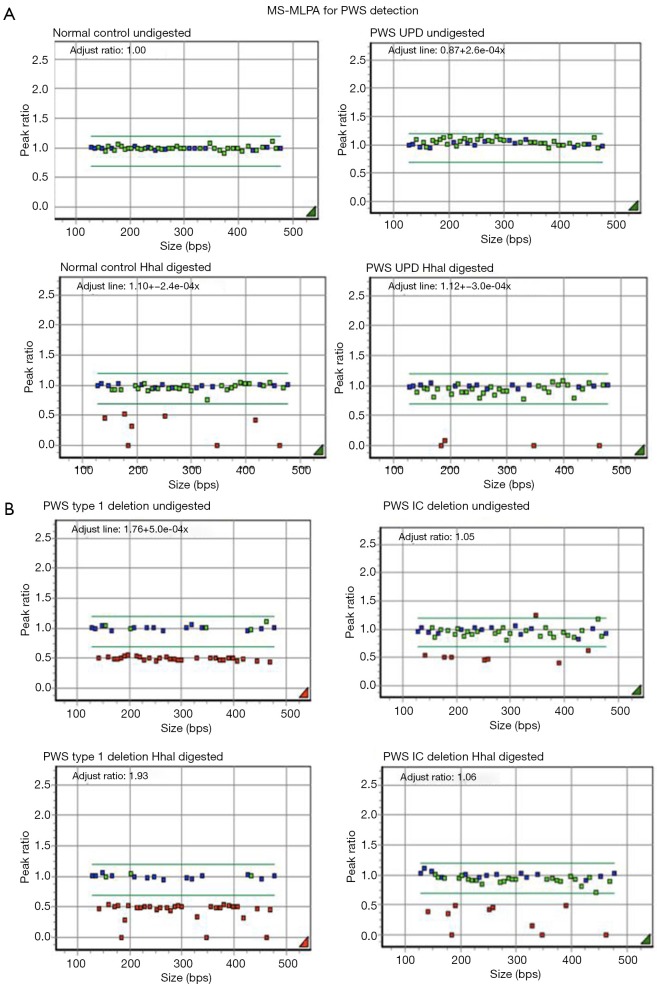
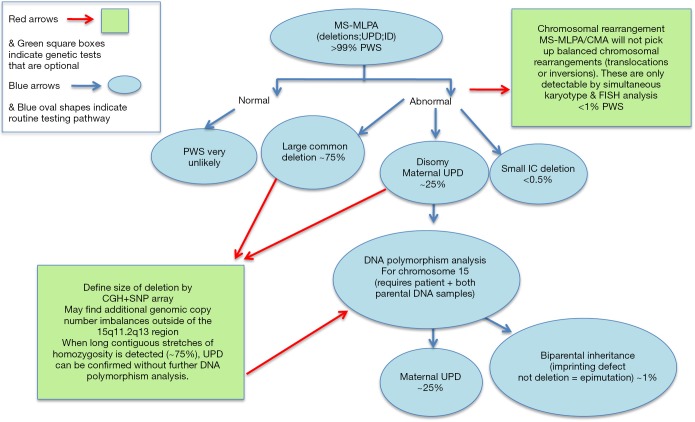
Although Prader-Willi syndrome (PWS) is a well-described clinical dysmorphic syndrome, DNA testing is required for a definitive diagnosis. A definitive diagnosis can be made in approximately 99% of cases using DNA testing; there are a number of DNA tests that can be used for this purpose, although there is no set standard algorithm of testing. The dilemma arises because of the complex genetic mechanisms at the basis of PWS, which need to be elucidated. To establish the molecular mechanism with a complete work up, involves at least 2 tests. Here we discuss the commonly used tests currently available and suggest a cost-effective approach to diagnostic testing.
Keywords: Cytogenetics; chromosome microarray; methylation.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Figures
Similar articles
-
Diagnostic and prognostic problems with the Prader-Willi syndrome.Proc (Bayl Univ Med Cent). 2019 Jan 11;32(1):167-168. doi: 10.1080/08998280.2018.1542477. eCollection 2019 Jan. Proc (Bayl Univ Med Cent). 2019. PMID: 30956621 Free PMC article.
-
The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria.Pediatrics. 2001 Nov;108(5):E92. doi: 10.1542/peds.108.5.e92. Pediatrics. 2001. PMID: 11694676
-
Molecular diagnosis of Prader-Willi syndrome.J Med Assoc Thai. 2003 Aug;86 Suppl 3:S510-6. J Med Assoc Thai. 2003. PMID: 14700141
-
Genetics of Prader-Willi syndrome and Prader-Will-Like syndrome.Ann Pediatr Endocrinol Metab. 2016 Sep;21(3):126-135. doi: 10.6065/apem.2016.21.3.126. Epub 2016 Sep 30. Ann Pediatr Endocrinol Metab. 2016. PMID: 27777904 Free PMC article. Review.
-
Genotype-Phenotype Relationships and Endocrine Findings in Prader-Willi Syndrome.Front Endocrinol (Lausanne). 2019 Dec 13;10:864. doi: 10.3389/fendo.2019.00864. eCollection 2019. Front Endocrinol (Lausanne). 2019. PMID: 31920975 Free PMC article. Review.
Cited by
-
Progress in Brain Magnetic Resonance Imaging of Individuals with Prader-Willi Syndrome.J Clin Med. 2023 Jan 29;12(3):1054. doi: 10.3390/jcm12031054. J Clin Med. 2023. PMID: 36769704 Free PMC article. Review.
-
Diagnostic and prognostic problems with the Prader-Willi syndrome.Proc (Bayl Univ Med Cent). 2019 Jan 11;32(1):167-168. doi: 10.1080/08998280.2018.1542477. eCollection 2019 Jan. Proc (Bayl Univ Med Cent). 2019. PMID: 30956621 Free PMC article.
-
Exploring the Diagnostic Complexity of Diabetes Subtypes in Pediatric Obesity: A Case Report of an Adolescent With Prader-Willi Phenotype and Literature Review.Cureus. 2024 Aug 8;16(8):e66456. doi: 10.7759/cureus.66456. eCollection 2024 Aug. Cureus. 2024. PMID: 39135667 Free PMC article.
-
Prader Willi Syndrome - A Common Epigenetic Cause of Syndromic Obesity.Indian J Pediatr. 2017 Nov;84(11):809-810. doi: 10.1007/s12098-017-2512-0. Epub 2017 Oct 2. Indian J Pediatr. 2017. PMID: 28971315 No abstract available.
-
Expanding deep phenotypic spectrum associated with atypical pathogenic structural variations overlapping 15q11-q13 imprinting region.Brain Behav. 2024 Apr;14(4):e3437. doi: 10.1002/brb3.3437. Brain Behav. 2024. PMID: 38616334 Free PMC article.
References
-
- Zellweger H. Differential Diagnosis in Prader-Willi Syndrome. In: Greenswag LR, Alexander RC, editors. Management of Prader-Willi Syndrome. Springer US;1988:15-22.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources