

Expanding Proteome Coverage with CHarge Ordered Parallel Ion aNalysis (CHOPIN) Combined with Broad Specificity Proteolysis
- PMID: 28164708
- PMCID: PMC5363888
- DOI: 10.1021/acs.jproteome.6b00915
Expanding Proteome Coverage with CHarge Ordered Parallel Ion aNalysis (CHOPIN) Combined with Broad Specificity Proteolysis
Abstract
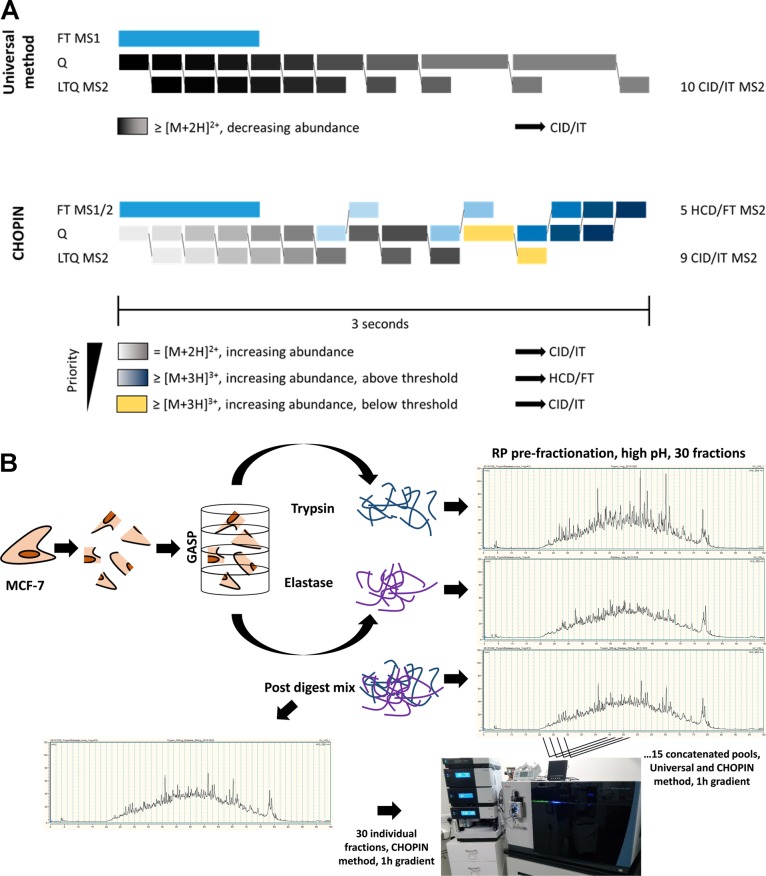
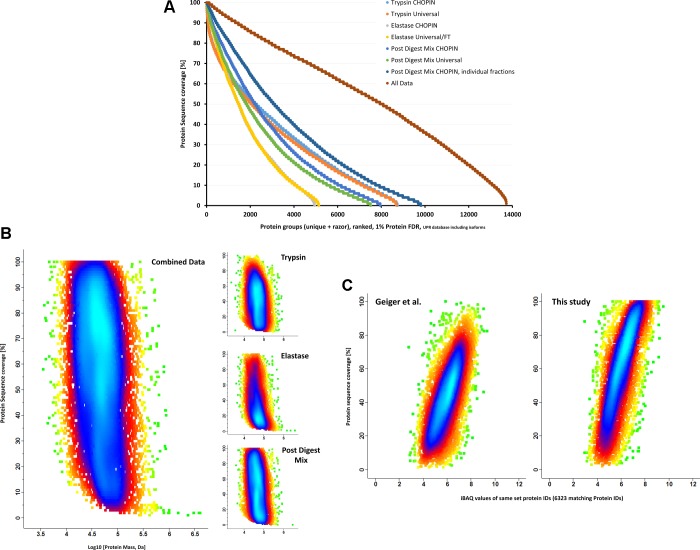
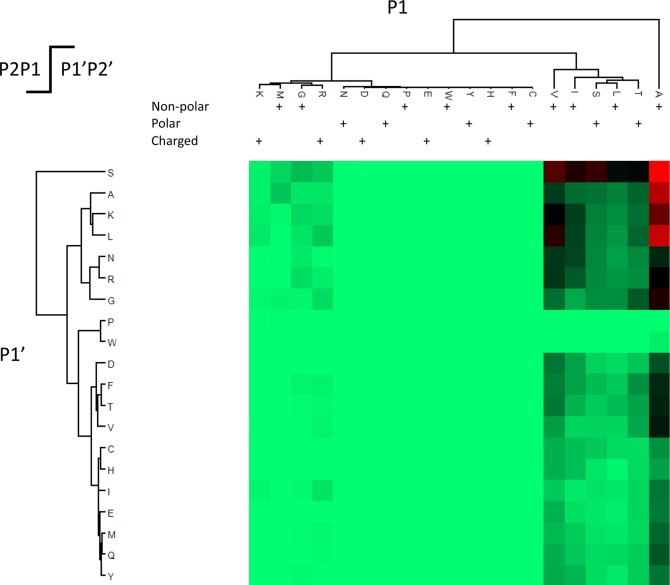
The "deep" proteome has been accessible by mass spectrometry for some time. However, the number of proteins identified in cells of the same type has plateaued at ∼8000-10 000 without ID transfer from reference proteomes/data. Moreover, limited sequence coverage hampers the discrimination of protein isoforms when using trypsin as standard protease. Multienzyme approaches appear to improve sequence coverage and subsequent isoform discrimination. Here we expanded proteome and protein sequence coverage in MCF-7 breast cancer cells to an as yet unmatched depth by employing a workflow that addresses current limitations in deep proteome analysis in multiple stages: We used (i) gel-aided sample preparation (GASP) and combined trypsin/elastase digests to increase peptide orthogonality, (ii) concatenated high-pH prefractionation, and (iii) CHarge Ordered Parallel Ion aNalysis (CHOPIN), available on an Orbitrap Fusion (Lumos) mass spectrometer, to achieve 57% median protein sequence coverage in 13 728 protein groups (8949 Unigene IDs) in a single cell line. CHOPIN allows the use of both detectors in the Orbitrap on predefined precursor types that optimizes parallel ion processing, leading to the identification of a total of 179 549 unique peptides covering the deep proteome in unprecedented detail.
Keywords: LC−MS/MS; deep proteome; isoform profiling; protein sequence coverage; sequence coverage.
Conflict of interest statement
The authors declare no competing financial interest.
Figures

References
-
- Wilhelm M.; Schlegl J.; Hahne H.; Moghaddas Gholami A.; Lieberenz M.; Savitski M. M.; Ziegler E.; Butzmann L.; Gessulat S.; Marx H.; Mathieson T.; Lemeer S.; Schnatbaum K.; Reimer U.; Wenschuh H.; Mollenhauer M.; Slotta-Huspenina J.; Boese J. H.; Bantscheff M.; Gerstmair A.; Faerber F.; Kuster B. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509 (7502), 582–7. 10.1038/nature13319. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
