

Integration of VDR genome wide binding and GWAS genetic variation data reveals co-occurrence of VDR and NF-κB binding that is linked to immune phenotypes
- PMID: 28166722
- PMCID: PMC5294817
- DOI: 10.1186/s12864-017-3481-4
Integration of VDR genome wide binding and GWAS genetic variation data reveals co-occurrence of VDR and NF-κB binding that is linked to immune phenotypes
Abstract
Background: The nuclear hormone receptor superfamily acts as a genomic sensor of diverse signals. Their actions are often intertwined with other transcription factors. Nuclear hormone receptors are targets for many therapeutic drugs, and include the vitamin D receptor (VDR). VDR signaling is pleotropic, being implicated in calcaemic function, antibacterial actions, growth control, immunomodulation and anti-cancer actions. Specifically, we hypothesized that the biologically significant relationships between the VDR transcriptome and phenotype-associated biology could be discovered by integrating the known VDR transcription factor binding sites and all published trait- and disease-associated SNPs. By integrating VDR genome-wide binding data (ChIP-seq) with the National Human Genome Research Institute (NHGRI) GWAS catalog of SNPs we would see where and which target gene interactions and pathways are impacted by inherited genetic variation in VDR binding sites, indicating which of VDR's multiple functions are most biologically significant.
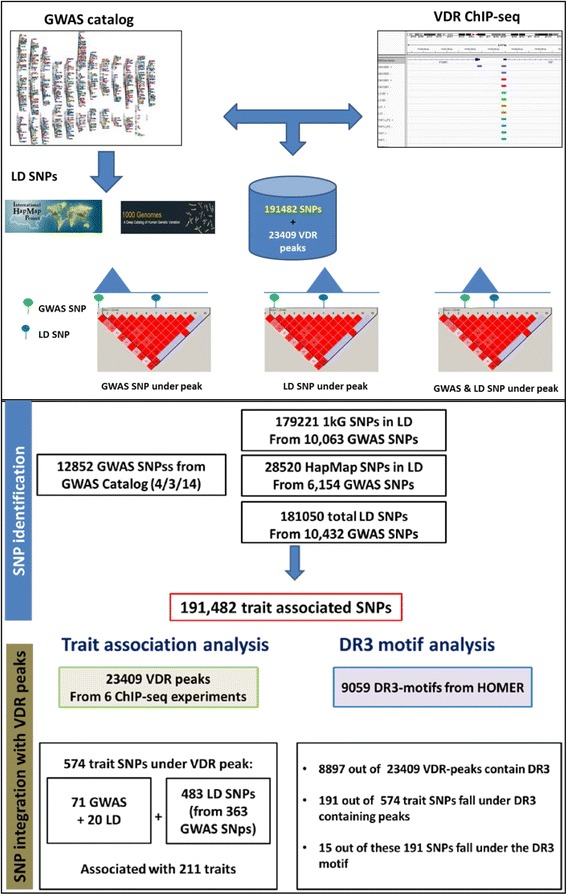
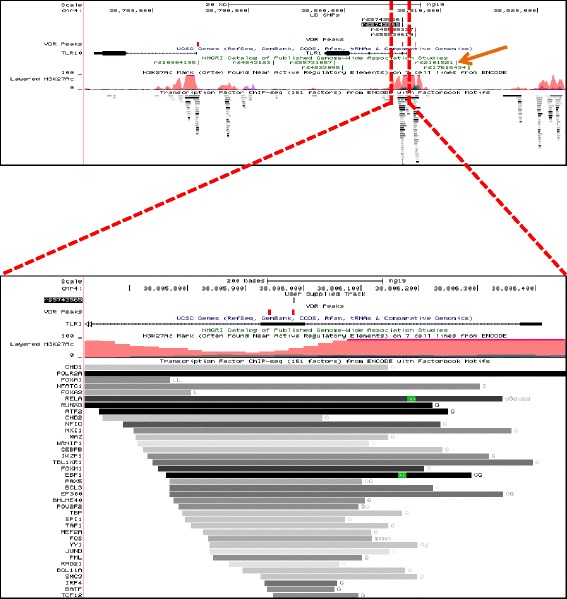
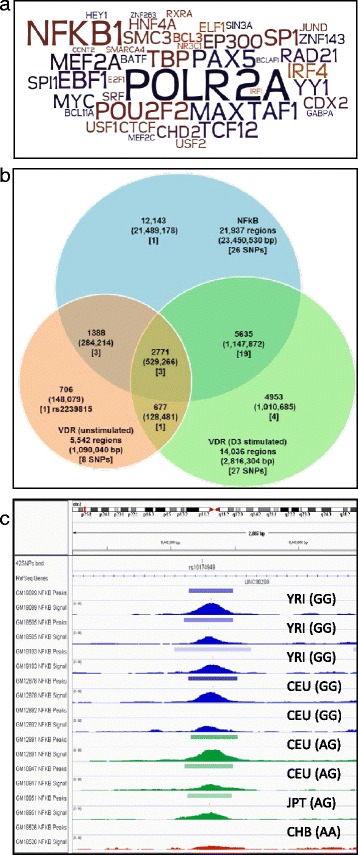
Results: To examine how genetic variation impacts VDR function we overlapped 23,409 VDR genomic binding peaks from six VDR ChIP-seq datasets with 191,482 SNPs, derived from GWAS-significant SNPs (Lead SNPs) and their correlated variants (r 2 > 0.8) from HapMap3 and the 1000 genomes project. In total, 574 SNPs (71 Lead and 503 SNPs in linkage disequilibrium with Lead SNPs) were present at VDR binding loci and associated with 211 phenotypes. For each phenotype a hypergeometric test was used to determine if SNPs were enriched at VDR binding sites. Bonferroni correction for multiple testing across the 211 phenotypes yielded 42 SNPs that were either disease- or phenotype-associated with seven predominately immune related including self-reported allergy; esophageal cancer was the only cancer phenotype. Motif analyses revealed that only two of these 42 SNPs reside within a canonical VDR binding site (DR3 motif), and that 1/3 of the 42 SNPs significantly impacted binding and gene regulation by other transcription factors, including NF-κB. This suggests a plausible link for the potential cross-talk between VDR and NF-κB.
Conclusions: These analyses showed that VDR peaks are enriched for SNPs associated with immune phenotypes suggesting that VDR immunomodulatory functions are amongst its most important actions. The enrichment of genetic variation in non-DR3 motifs suggests a significant role for the VDR to bind in multimeric complexes containing other transcription factors that are the primary DNA binding component. Our work provides a framework for the combination of ChIP-seq and GWAS findings to provide insight into the underlying phenotype-associated biology of a given transcription factor.
Keywords: ChIP-seq; Cistrome; DR3 motif; GWAS; Immune function; Linkage disequilibrium; NF-κB; Nuclear receptor superfamily; SNP; VDR.
Figures
Similar articles
-
Dynamics of 1α,25-dihydroxyvitamin D3-dependent chromatin accessibility of early vitamin D receptor target genes.Biochim Biophys Acta. 2013 Dec;1829(12):1266-75. doi: 10.1016/j.bbagrm.2013.10.003. Epub 2013 Nov 1. Biochim Biophys Acta. 2013. PMID: 24185200
-
Identification of genetic variants affecting vitamin D receptor binding and associations with autoimmune disease.Hum Mol Genet. 2017 Jun 1;26(11):2164-2176. doi: 10.1093/hmg/ddx092. Hum Mol Genet. 2017. PMID: 28335003 Free PMC article.
-
Patterns of genome-wide VDR locations.PLoS One. 2014 Apr 30;9(4):e96105. doi: 10.1371/journal.pone.0096105. eCollection 2014. PLoS One. 2014. PMID: 24787735 Free PMC article.
-
Bioinformatic approaches to interrogating vitamin D receptor signaling.Mol Cell Endocrinol. 2017 Sep 15;453:3-13. doi: 10.1016/j.mce.2017.03.011. Epub 2017 Mar 10. Mol Cell Endocrinol. 2017. PMID: 28288905 Review.
-
Molecular endocrinology of vitamin D on the epigenome level.Mol Cell Endocrinol. 2017 Sep 15;453:14-21. doi: 10.1016/j.mce.2017.03.016. Epub 2017 Mar 16. Mol Cell Endocrinol. 2017. PMID: 28315703 Review.
Cited by
-
Nuclear receptors in cancer - uncovering new and evolving roles through genomic analysis.Nat Rev Genet. 2018 Mar;19(3):160-174. doi: 10.1038/nrg.2017.102. Epub 2017 Dec 27. Nat Rev Genet. 2018. PMID: 29279606 Review.
-
Influence of Vitamin D on Islet Autoimmunity and Beta-Cell Function in Type 1 Diabetes.Nutrients. 2019 Sep 11;11(9):2185. doi: 10.3390/nu11092185. Nutrients. 2019. PMID: 31514368 Free PMC article. Review.
-
Ancient Nuclear Receptor VDR With New Functions: Microbiome and Inflammation.Inflamm Bowel Dis. 2018 May 18;24(6):1149-1154. doi: 10.1093/ibd/izy092. Inflamm Bowel Dis. 2018. PMID: 29718408 Free PMC article. Review.
-
High Levels of Circulating Type II Collagen Degradation Marker (CTx-II) Are Associated with Specific VDR Polymorphisms in Patients with Adult Vertebral Osteochondrosis.Int J Mol Sci. 2017 Sep 29;18(10):2073. doi: 10.3390/ijms18102073. Int J Mol Sci. 2017. PMID: 28961166 Free PMC article.
-
Nongenomic Activities of Vitamin D.Nutrients. 2022 Dec 1;14(23):5104. doi: 10.3390/nu14235104. Nutrients. 2022. PMID: 36501134 Free PMC article. Review.
References
-
- Consortium EP, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447(7146):799–816. doi: 10.1038/nature05874. - DOI - PMC - PubMed
-
- Kawaji H, Severin J, Lizio M, Forrest AR, van Nimwegen E, Rehli M, Schroder K, Irvine K, Suzuki H, Carninci P, et al. Update of the FANTOM web resource: from mammalian transcriptional landscape to its dynamic regulation. Nucleic Acids Res. 2011;39(Database issue):D856–60. doi: 10.1093/nar/gkq1112. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
