

Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation
- PMID: 28166811
- PMCID: PMC5295186
- DOI: 10.1186/s13073-017-0403-7
Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation
Abstract
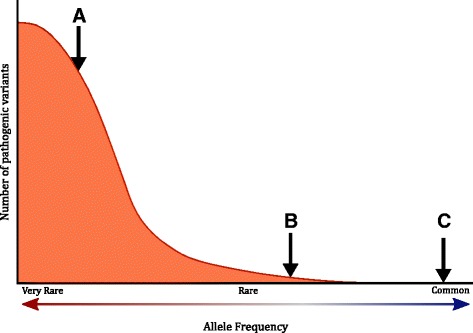
Background: The frequency of a variant in the general population is a key criterion used in the clinical interpretation of sequence variants. With certain exceptions, such as founder mutations, the rarity of a variant is a prerequisite for pathogenicity. However, defining the threshold at which a variant should be considered "too common" is challenging and therefore diagnostic laboratories have typically set conservative allele frequency thresholds.
Methods: Recent publications of large population sequencing data, such as the Exome Aggregation Consortium (ExAC) database, provide an opportunity to characterize with accuracy and precision the frequency distributions of very rare disease-causing alleles. Allele frequencies of pathogenic variants in ClinVar, as well as variants expected to be pathogenic through the nonsense-mediated decay (NMD) pathway, were analyzed to study the burden of pathogenic variants in 79 genes of clinical importance.
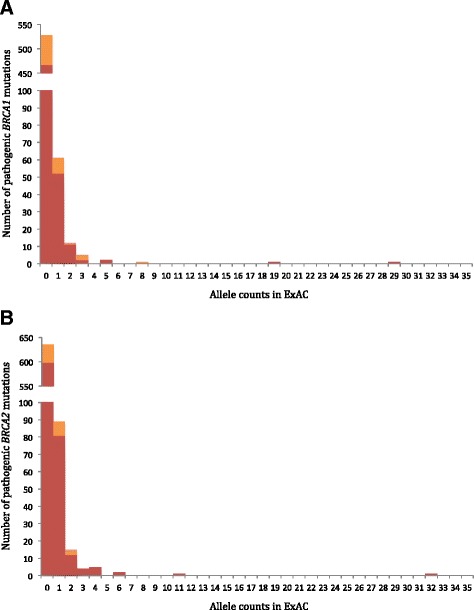
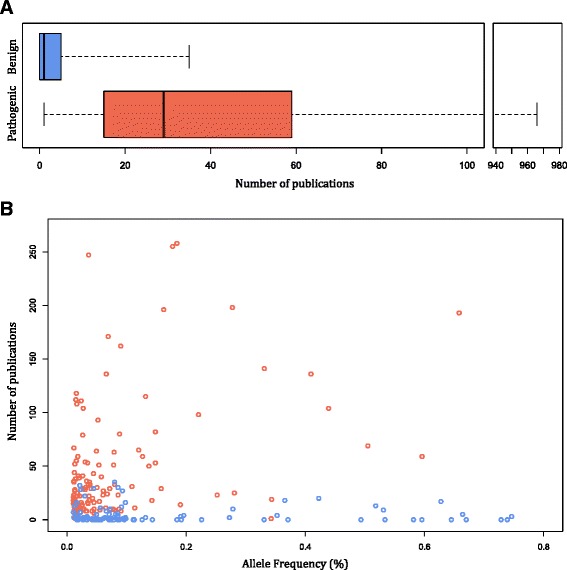
Results: Of 1364 BRCA1 and BRCA2 variants that are well characterized as pathogenic or that are expected to lead to NMD, 1350 variants had an allele frequency of less than 0.0025%. The remaining 14 variants were previously published founder mutations. Importantly, we observed no difference in the distributions of pathogenic variants expected to be lead to NMD compared to those that are not. Therefore, we expanded the analysis to examine the distributions of NMD expected variants in 77 additional genes. These 77 genes were selected to represent a broad set of clinical areas, modes of inheritance, and penetrance. Among these variants, most (97.3%) had an allele frequency of less than 0.01%. Furthermore, pathogenic variants with allele frequencies greater than 0.01% were well characterized in publications and included many founder mutations.
Conclusions: The observations made in this study suggest that, with certain caveats, a very low allele frequency threshold can be adopted to more accurately interpret sequence variants.
Keywords: ACMG ISV guidelines; Allele-frequency threshold; ExAC; Variant interpretation.
Figures
References
-
- ClinVar. 2016. http://www.ncbi.nlm.nih.gov/clinvar/. Accessed 28 June 2016.
-
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
