

Microbial strain-level population structure and genetic diversity from metagenomes
- PMID: 28167665
- PMCID: PMC5378180
- DOI: 10.1101/gr.216242.116
Microbial strain-level population structure and genetic diversity from metagenomes
Abstract
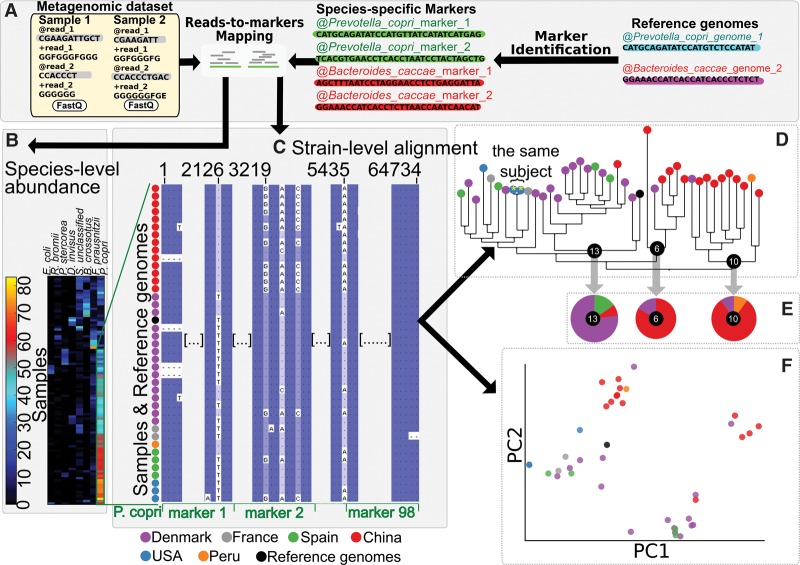
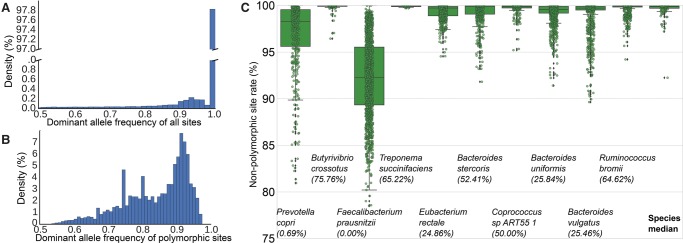
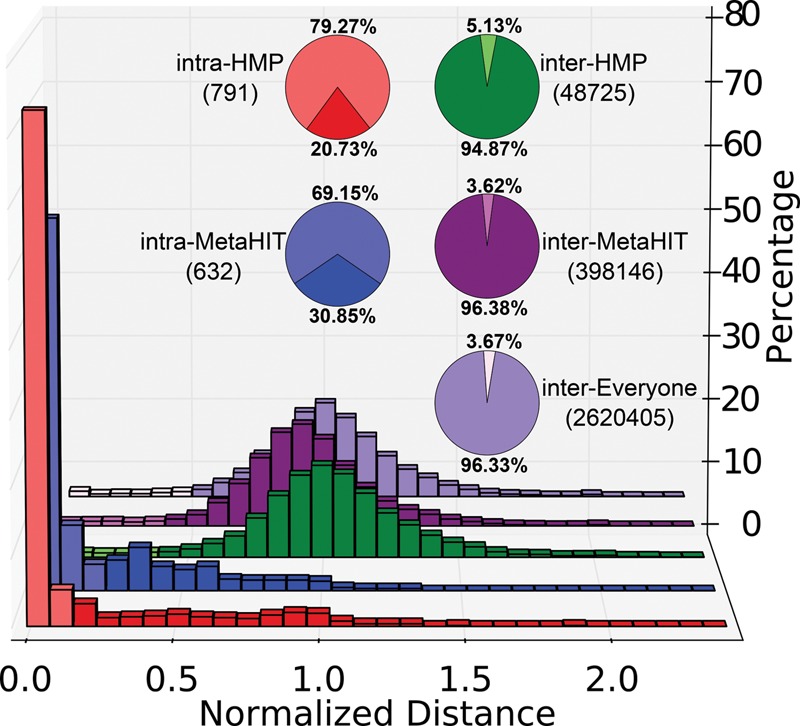
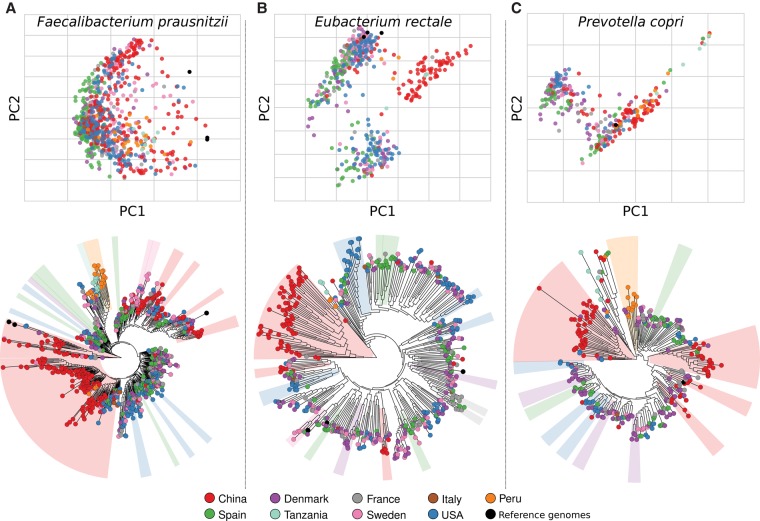
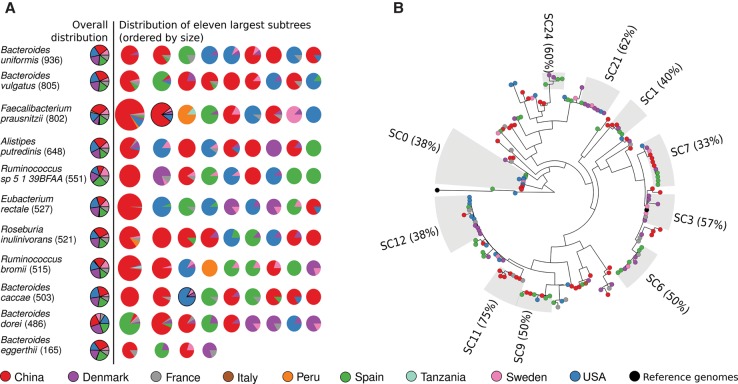
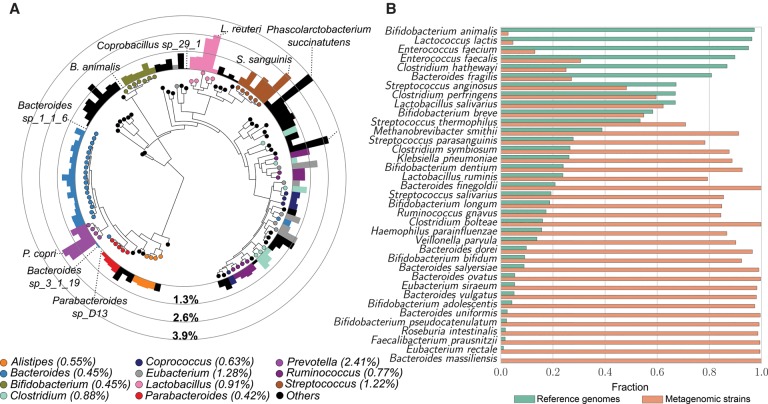
Among the human health conditions linked to microbial communities, phenotypes are often associated with only a subset of strains within causal microbial groups. Although it has been critical for decades in microbial physiology to characterize individual strains, this has been challenging when using culture-independent high-throughput metagenomics. We introduce StrainPhlAn, a novel metagenomic strain identification approach, and apply it to characterize the genetic structure of thousands of strains from more than 125 species in more than 1500 gut metagenomes drawn from populations spanning North and South American, European, Asian, and African countries. The method relies on per-sample dominant sequence variant reconstruction within species-specific marker genes. It identified primarily subject-specific strain variants (<5% inter-subject strain sharing), and we determined that a single strain typically dominated each species and was retained over time (for >70% of species). Microbial population structure was correlated in several distinct ways with the geographic structure of the host population. In some cases, discrete subspecies (e.g., for Eubacterium rectale and Prevotella copri) or continuous microbial genetic variations (e.g., for Faecalibacterium prausnitzii) were associated with geographically distinct human populations, whereas few strains occurred in multiple unrelated cohorts. We further estimated the genetic variability of gut microbes, with Bacteroides species appearing remarkably consistent (0.45% median number of nucleotide variants between strains), whereas P. copri was among the most plastic gut colonizers. We thus characterize here the population genetics of previously inaccessible intestinal microbes, providing a comprehensive strain-level genetic overview of the gut microbial diversity.
© 2017 Truong et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Achtman M, Wagner M. 2008. Microbial diversity and the genetic nature of microbial species. Nat Rev Microbiol 6: 431–440. - PubMed
-
- Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C. 2014. Binning metagenomic contigs by coverage and composition. Nat Methods 11: 1144–1146. - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources