

Conformational disruption of PI3Kδ regulation by immunodeficiency mutations in PIK3CD and PIK3R1
- PMID: 28167755
- PMCID: PMC5338455
- DOI: 10.1073/pnas.1617244114
Conformational disruption of PI3Kδ regulation by immunodeficiency mutations in PIK3CD and PIK3R1
Abstract
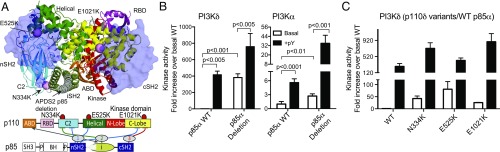
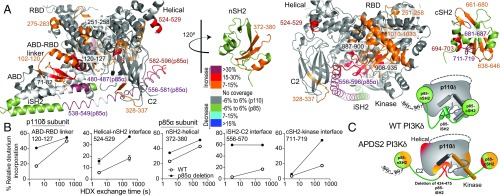
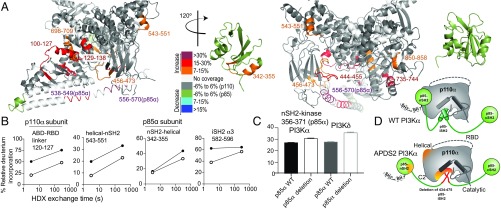
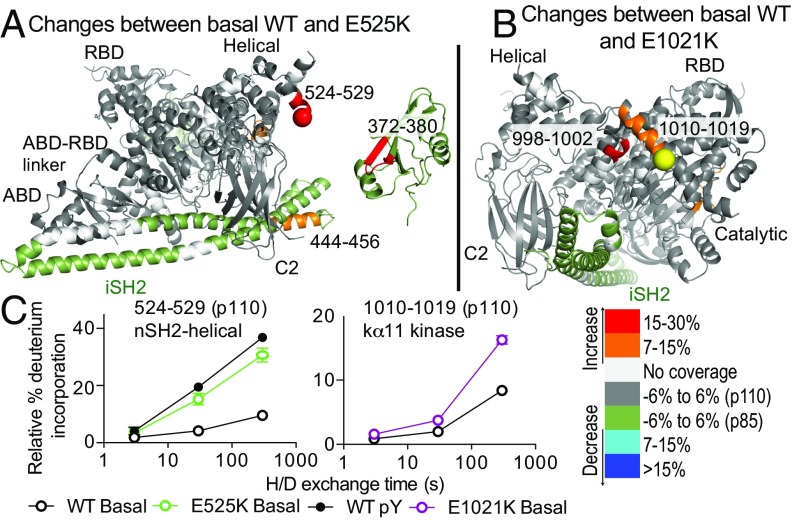
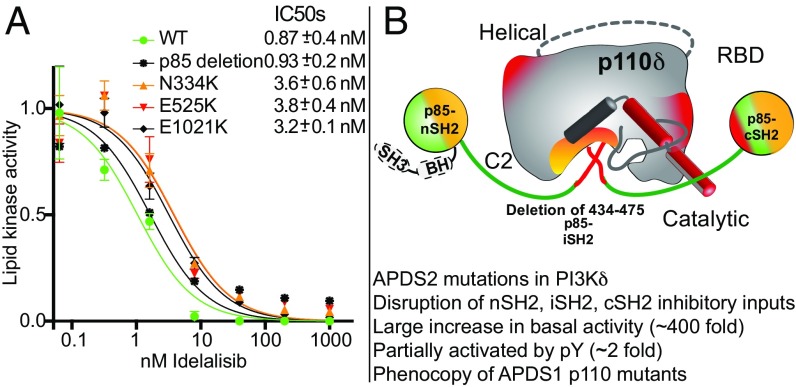
Activated PI3K Delta Syndrome (APDS) is a primary immunodeficiency disease caused by activating mutations in either the leukocyte-restricted p110δ catalytic (PIK3CD) subunit or the ubiquitously expressed p85α regulatory (PIK3R1) subunit of class IA phosphoinositide 3-kinases (PI3Ks). There are two classes of APDS: APDS1 that arises from p110δ mutations that are analogous to oncogenic mutations found in the broadly expressed p110α subunit and APDS2 that occurs from a splice mutation resulting in p85α with a central deletion (Δ434-475). As p85 regulatory subunits associate with and inhibit all class IA catalytic subunits, APDS2 mutations are expected to similarly activate p110α, β, and δ, yet APDS2 largely phenocopies APDS1 without dramatic effects outside the immune system. We have examined the molecular mechanism of activation of both classes of APDS mutations using a combination of biochemical assays and hydrogen-deuterium exchange mass spectrometry. Intriguingly, we find that an APDS2 mutation in p85α leads to substantial basal activation of p110δ (>300-fold) and disrupts inhibitory interactions from the nSH2, iSH2, and cSH2 domains of p85, whereas p110α is only minimally basally activated (∼2-fold) when associated with mutated p85α. APDS1 mutations in p110δ (N334K, E525K, E1021K) mimic the activation mechanisms previously discovered for oncogenic mutations in p110α. All APDS mutations were potently inhibited by the Food and Drug Administration-approved p110δ inhibitor idelalisib. Our results define the molecular basis of how PIK3CD and PIK3R1 mutations result in APDS and reveal a potential path to treatment for all APDS patients.
Keywords: HDX-MS; PI3K/AKT; PIK3CD; PIK3R1; phosphoinositides.
Conflict of interest statement
Conflict of interest statement: C.L.L. collaborates with Novartis on related studies.
Figures
References
-
- Burke JE, Williams RL. Synergy in activating class I PI3Ks. Trends Biochem Sci. 2015;40(2):88–100. - PubMed
-
- Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329–341. - PubMed
-
- Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
