

Cell population structure prior to bifurcation predicts efficiency of directed differentiation in human induced pluripotent cells
- PMID: 28167799
- PMCID: PMC5338498
- DOI: 10.1073/pnas.1621412114
Cell population structure prior to bifurcation predicts efficiency of directed differentiation in human induced pluripotent cells
Abstract
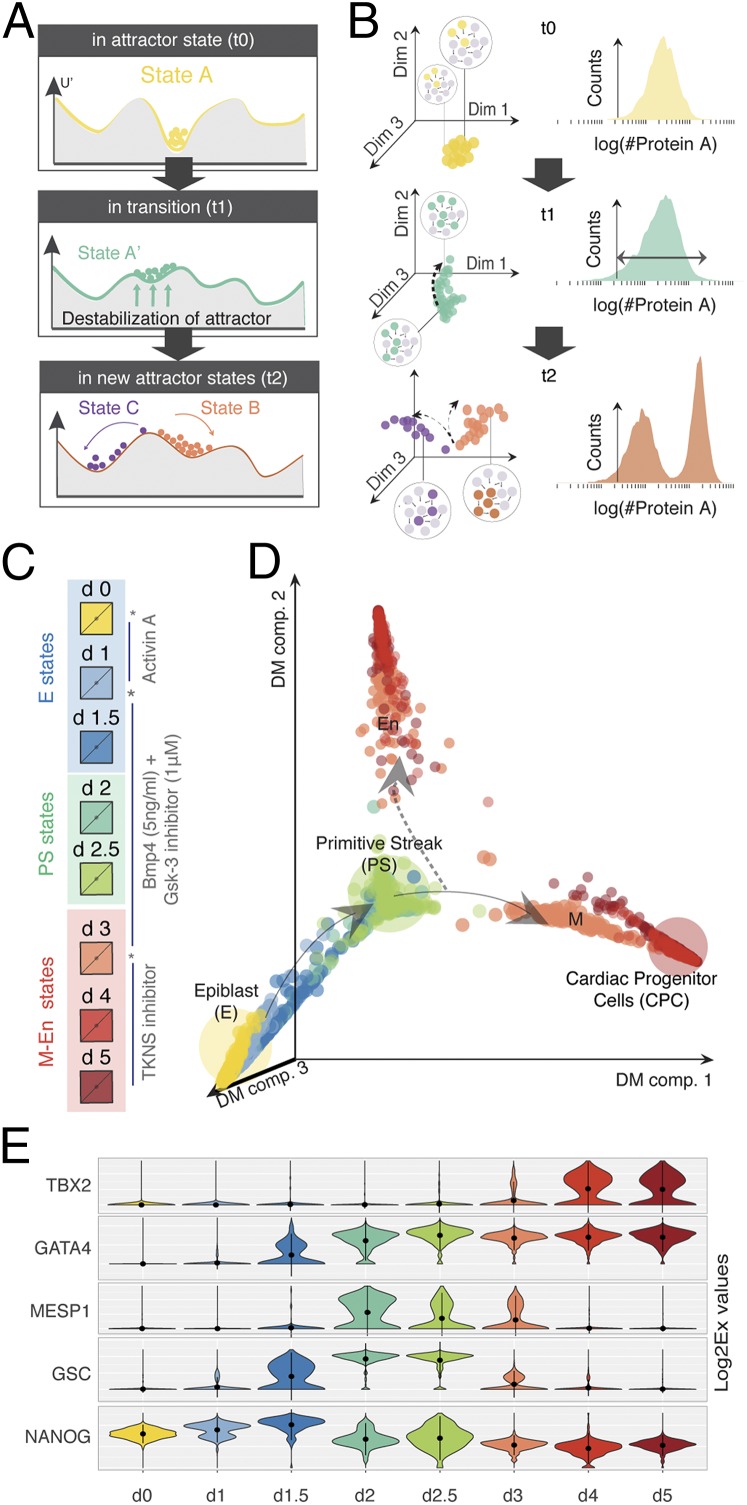
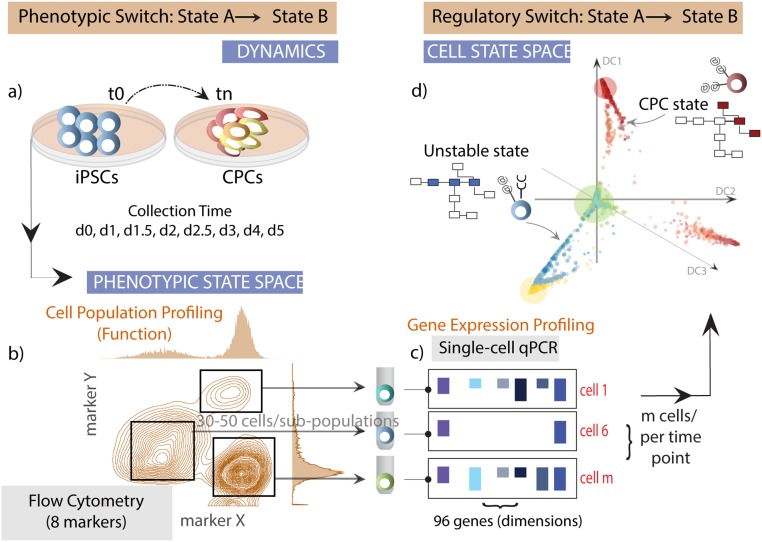
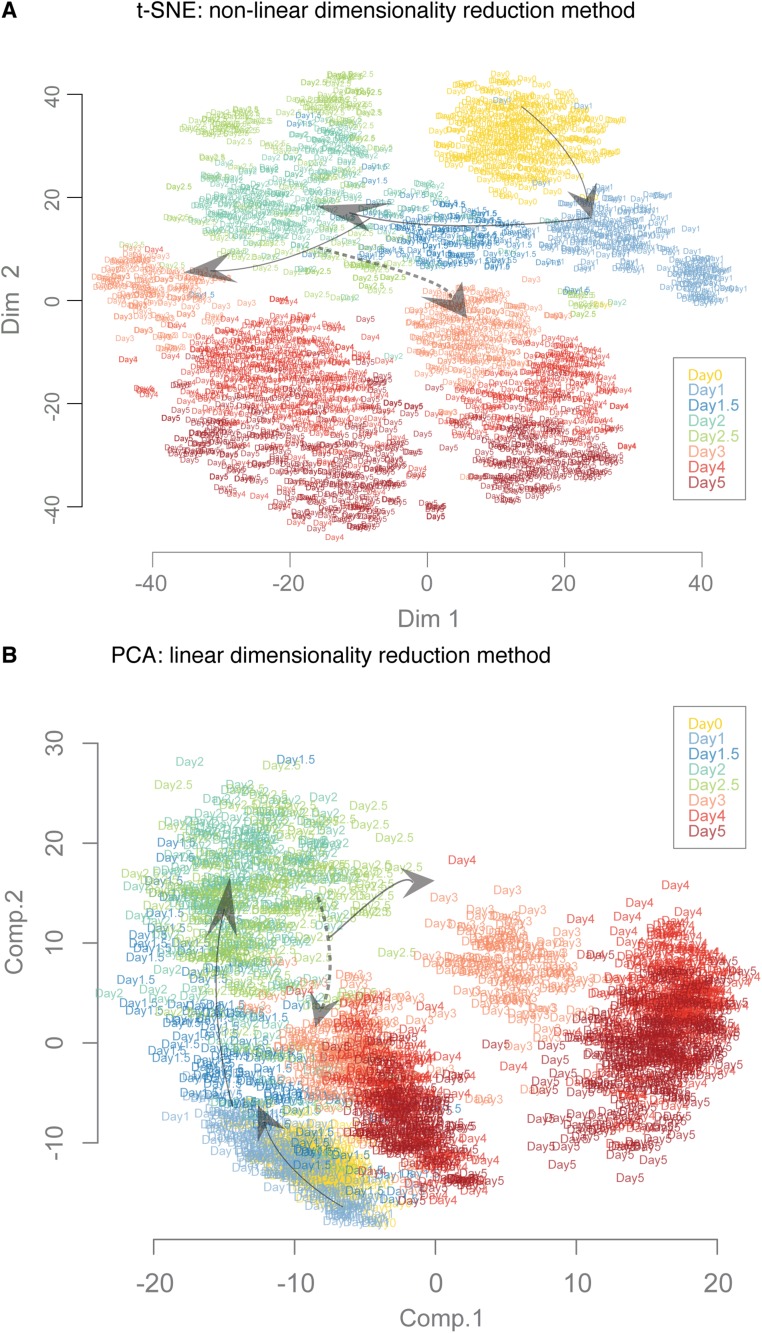
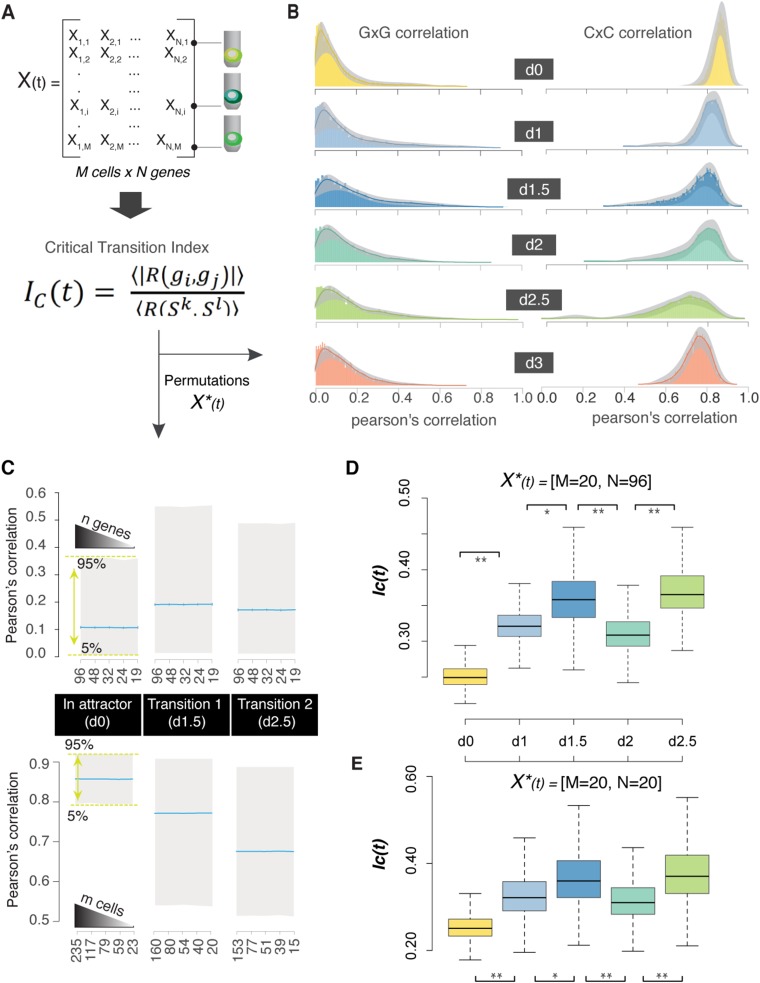
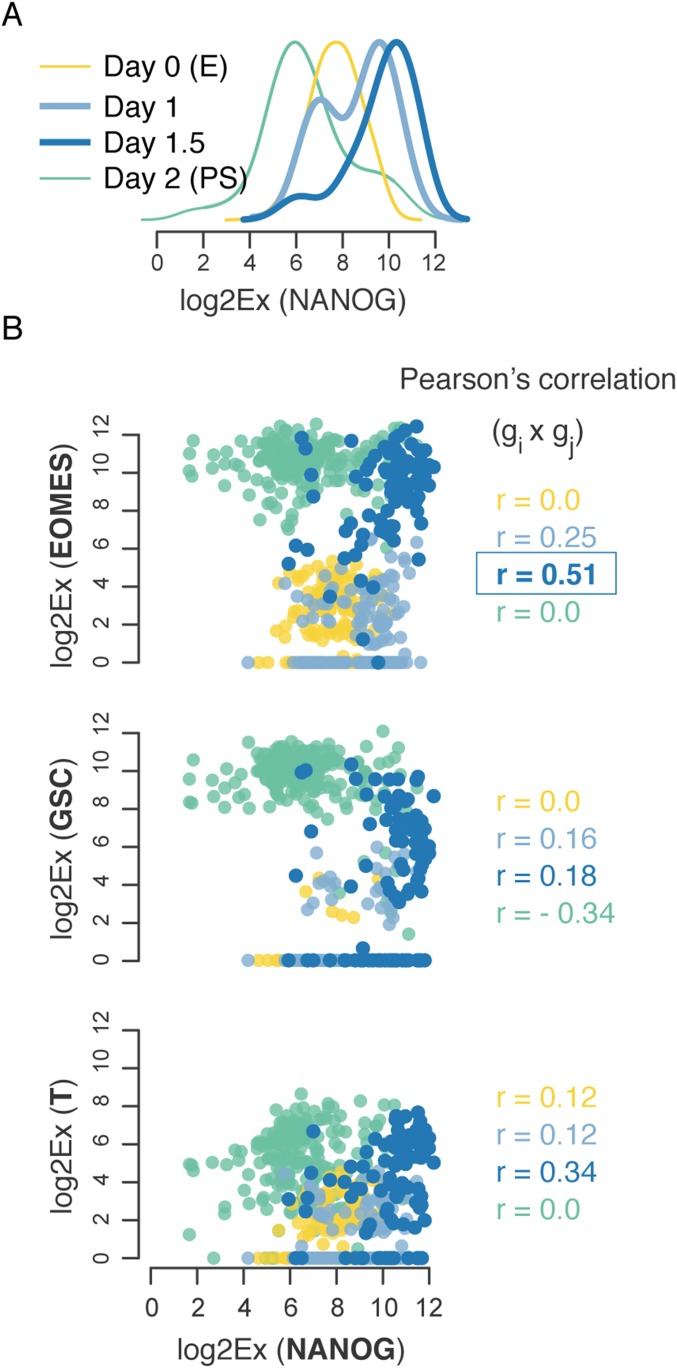
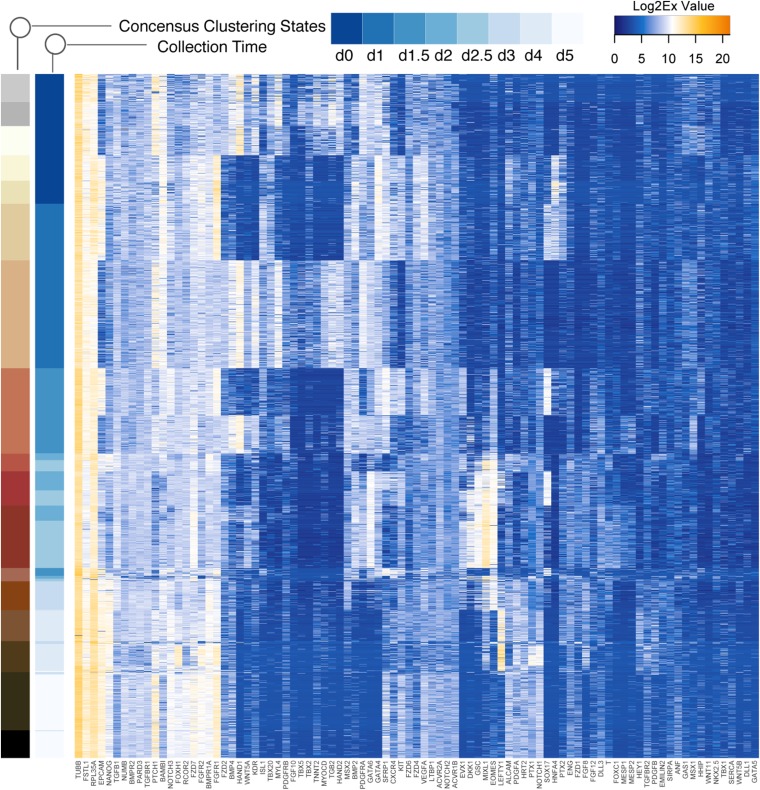
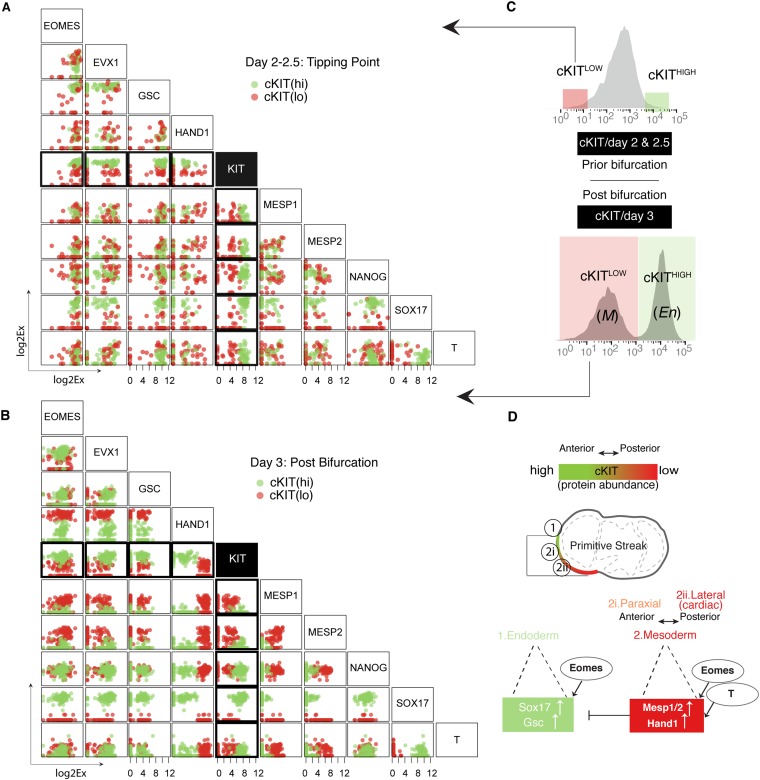
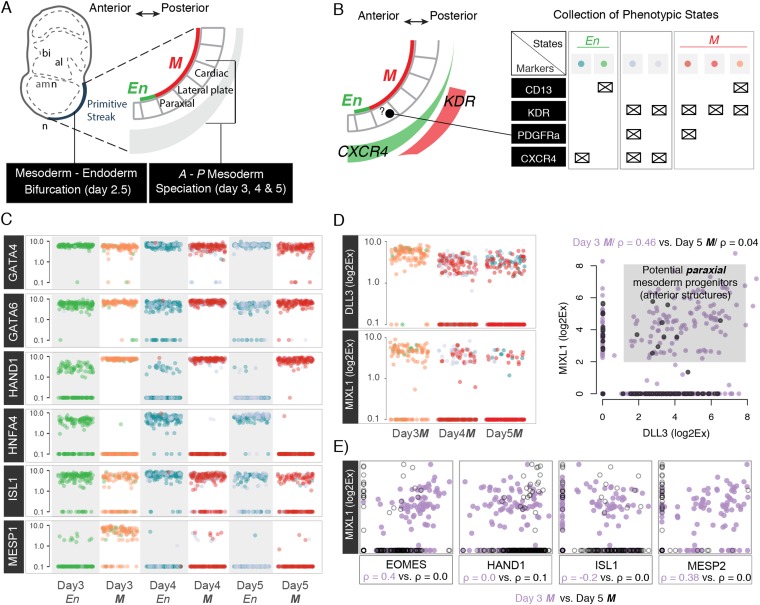
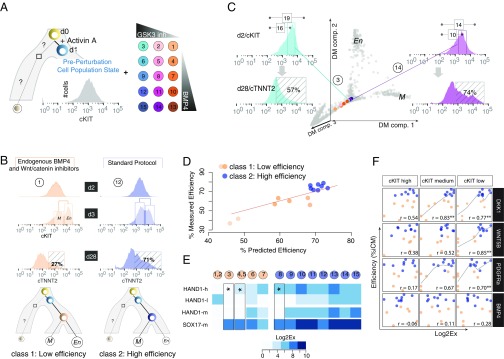
Steering the differentiation of induced pluripotent stem cells (iPSCs) toward specific cell types is crucial for patient-specific disease modeling and drug testing. This effort requires the capacity to predict and control when and how multipotent progenitor cells commit to the desired cell fate. Cell fate commitment represents a critical state transition or "tipping point" at which complex systems undergo a sudden qualitative shift. To characterize such transitions during iPSC to cardiomyocyte differentiation, we analyzed the gene expression patterns of 96 developmental genes at single-cell resolution. We identified a bifurcation event early in the trajectory when a primitive streak-like cell population segregated into the mesodermal and endodermal lineages. Before this branching point, we could detect the signature of an imminent critical transition: increase in cell heterogeneity and coordination of gene expression. Correlation analysis of gene expression profiles at the tipping point indicates transcription factors that drive the state transition toward each alternative cell fate and their relationships with specific phenotypic readouts. The latter helps us to facilitate small molecule screening for differentiation efficiency. To this end, we set up an analysis of cell population structure at the tipping point after systematic variation of the protocol to bias the differentiation toward mesodermal or endodermal cell lineage. We were able to predict the proportion of cardiomyocytes many days before cells manifest the differentiated phenotype. The analysis of cell populations undergoing a critical state transition thus affords a tool to forecast cell fate outcomes and can be used to optimize differentiation protocols to obtain desired cell populations.
Keywords: critical state transitions; differentiation efficiency; iPSC to cardiomyocyte differentiation; prediction; single-cell analysis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol. 2016;17(3):170–182. - PubMed
-
- Nelson TJ, Martinez-Fernandez A, Terzic A. Induced pluripotent stem cells: Developmental biology to regenerative medicine. Nat Rev Cardiol. 2010;7(12):700–710. - PubMed
-
- Semrau S, et al. 2016. Dynamics of lineage commitment revealed by single-cell transcriptomics of differentiating embryonic stem cells. bioRxiv:10.1101/068288.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
