

Germline and somatic BAP1 mutations in high-grade rhabdoid meningiomas
- PMID: 28170043
- PMCID: PMC5464371
- DOI: 10.1093/neuonc/now235
Germline and somatic BAP1 mutations in high-grade rhabdoid meningiomas
Abstract
Background: Patients with meningiomas have widely divergent clinical courses. Some entirely recover following surgery alone, while others have relentless tumor recurrences. This clinical conundrum is exemplified by rhabdoid meningiomas, which are designated in the World Health Organization Classification of Tumours as high grade, despite only a subset following an aggressive clinical course. Patient management decisions are further exacerbated by high rates of interobserver variability, biased against missing possibly aggressive tumors. Objective molecular determinants are needed to guide classification and clinical decision making.
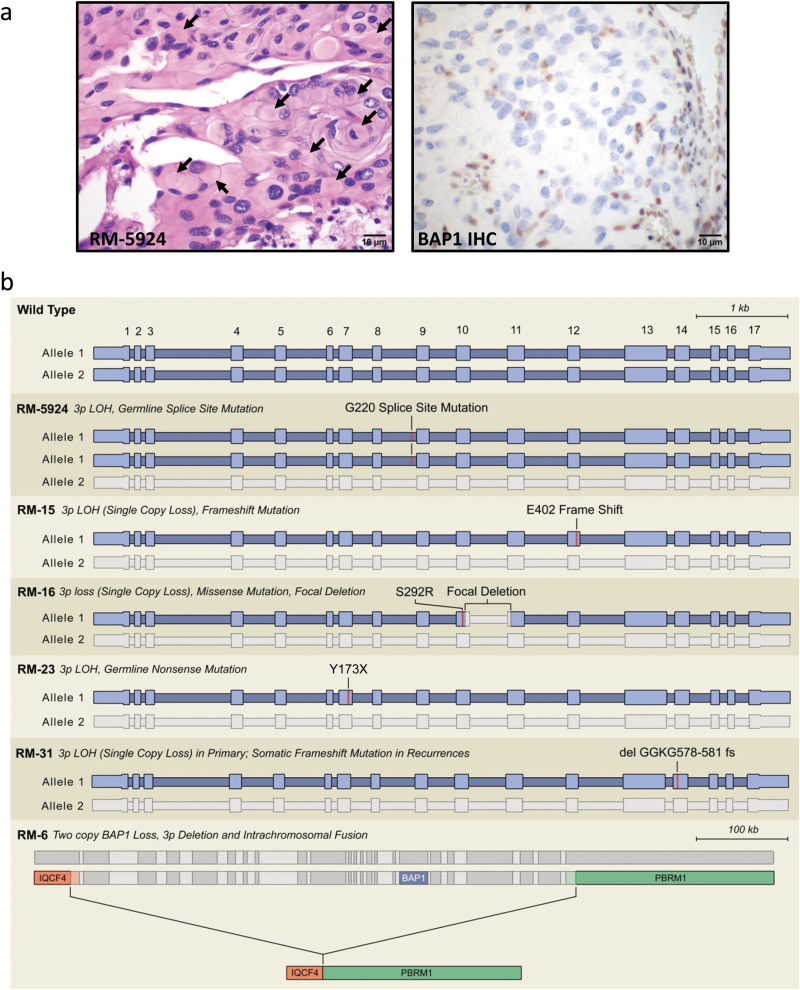
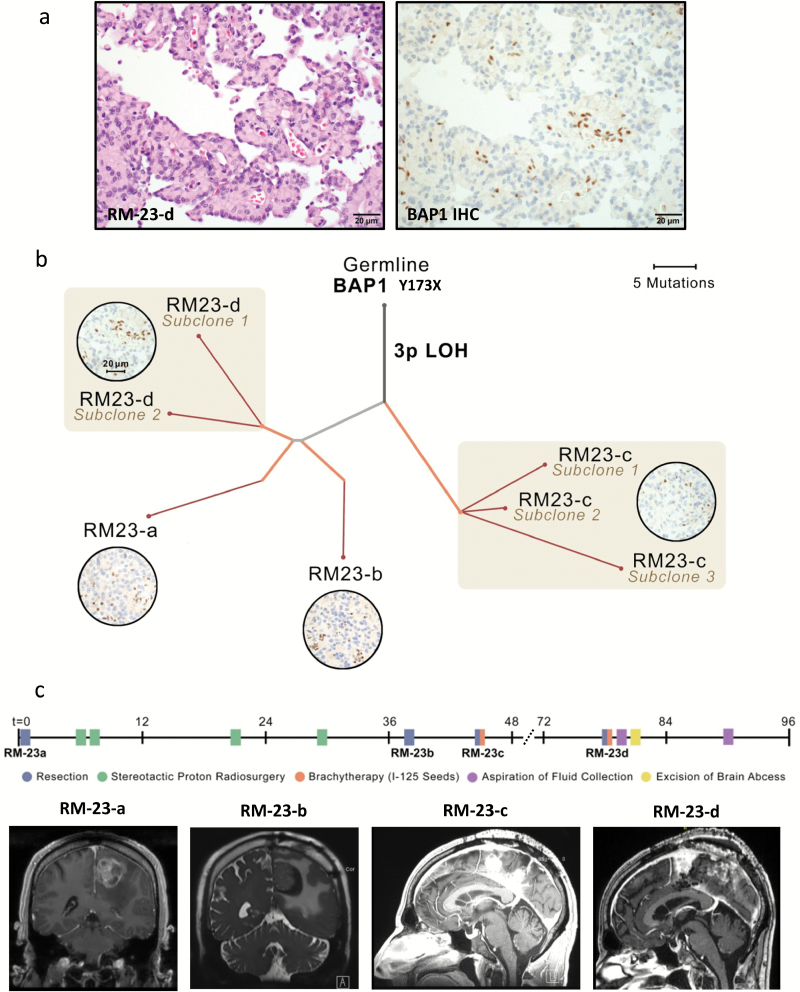
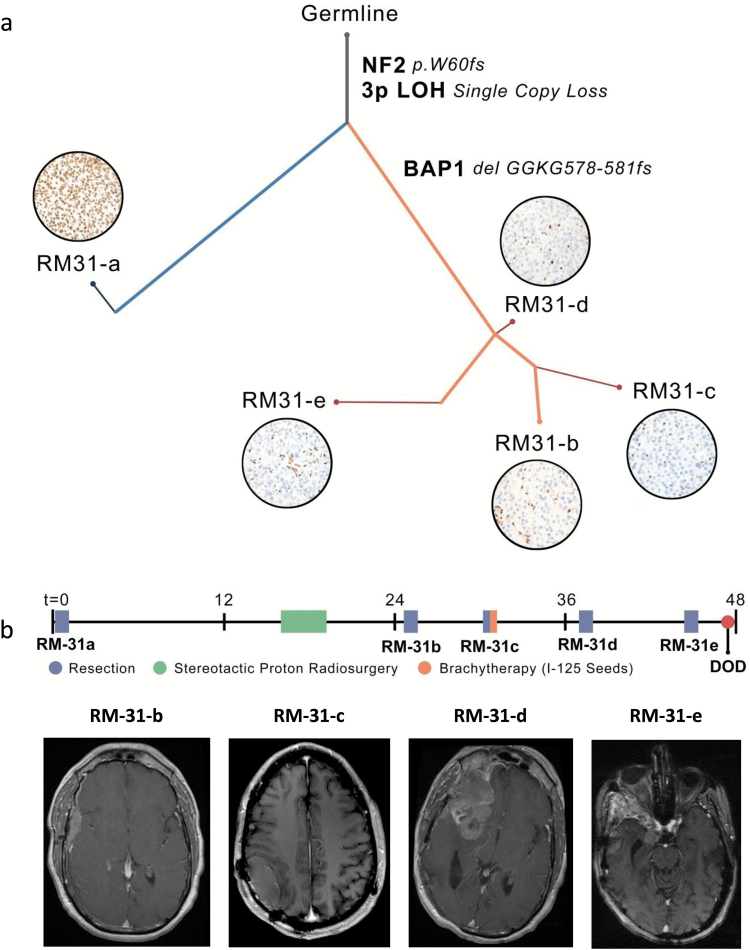
Methods: To define genomic aberrations of rhabdoid meningiomas, we performed sequencing of cancer-related genes in 27 meningiomas from 18 patients with rhabdoid features and evaluated breast cancer [BRCA]1-associated protein 1 (BAP1) expression by immunohistochemistry in 336 meningiomas. We assessed outcomes, germline status, and family history in patients with BAP1-negative rhabdoid meningiomas.
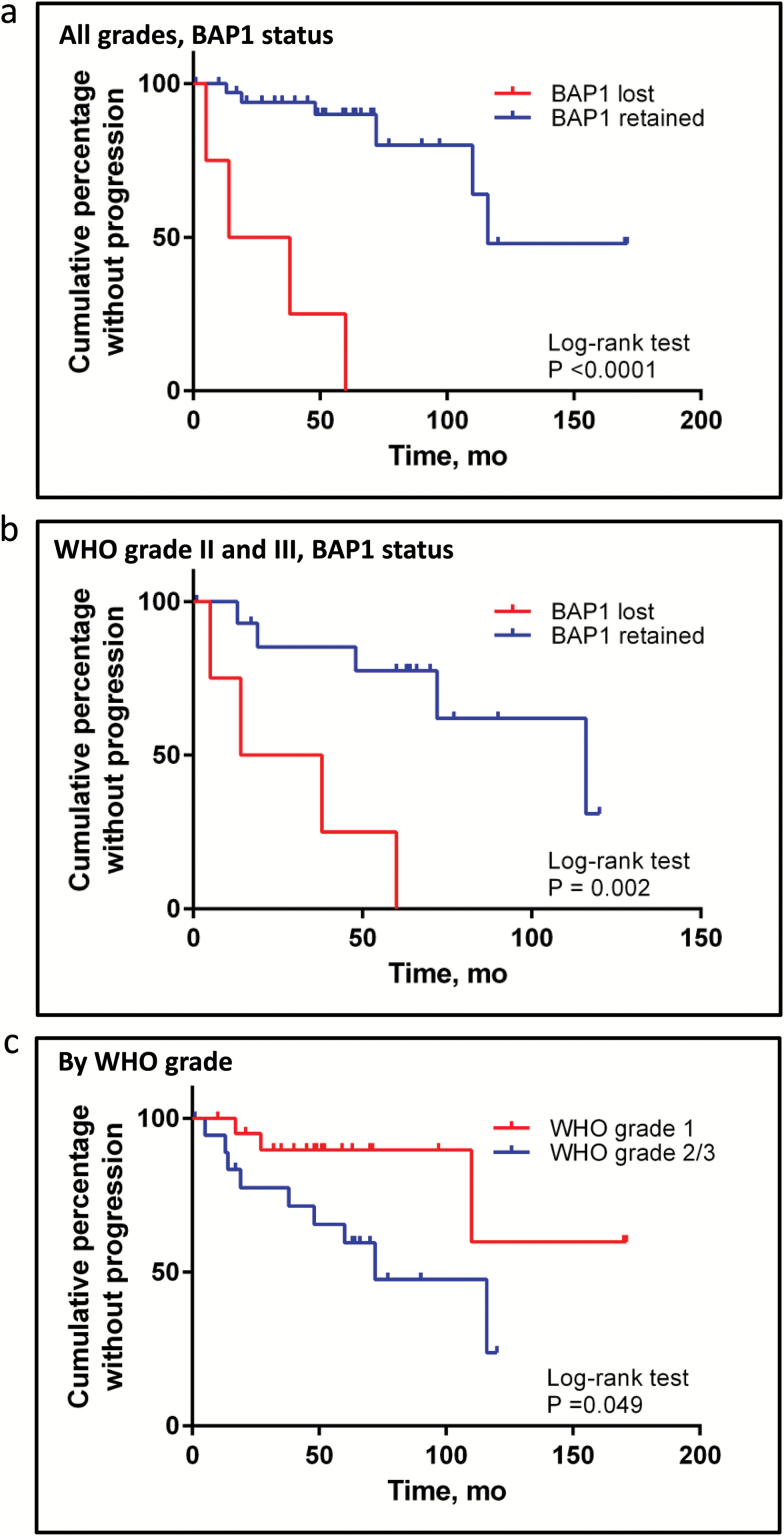
Results: The tumor suppressor gene BAP1, a ubiquitin carboxy-terminal hydrolase, is inactivated in a subset of high-grade rhabdoid meningiomas. Patients with BAP1-negative rhabdoid meningiomas had reduced time to recurrence compared with patients with BAP1-retained rhabdoid meningiomas (Kaplan-Meier analysis, 26 mo vs 116 mo, P < .001; hazard ratio 12.89). A subset of patients with BAP1-deficient rhabdoid meningiomas harbored germline BAP1 mutations, indicating that rhabdoid meningiomas can be a harbinger of the BAP1 cancer predisposition syndrome.
Conclusion: We define a subset of aggressive rhabdoid meningiomas that can be recognized using routine laboratory tests. We implicate ubiquitin deregulation in the pathogenesis of these high-grade malignancies. In addition, we show that familial and sporadic BAP1-mutated rhabdoid meningiomas are clinically aggressive, requiring intensive clinical management.
Keywords: BAP1; exome sequencing; rhabdoid meningiomas.
© The Author(s) 2016. Published by Oxford University Press on behalf of the Society for Neuro-Oncology. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com
Figures
References
-
- Louis DN, Ohgaki H, Wiestler OD, et al. WHO Classification of Tumours of the Central Nervous System. Revised 4th ed. Lyon, France: International Agency for Research on Cancer; 2016:232–257.
-
- Reuss DE, Piro RM, Jones DT, et al. Secretory meningiomas are defined by combined KLF4 K409Q and TRAF7 mutations. Acta Neuropathol. 2013;125(3):351–358. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
