

Emerging Concepts of Data Integration in Pathogen Phylodynamics
- PMID: 28173504
- PMCID: PMC5837209
- DOI: 10.1093/sysbio/syw054
Emerging Concepts of Data Integration in Pathogen Phylodynamics
Abstract
Phylodynamics has become an increasingly popular statistical framework to extract evolutionary and epidemiological information from pathogen genomes. By harnessing such information, epidemiologists aim to shed light on the spatio-temporal patterns of spread and to test hypotheses about the underlying interaction of evolutionary and ecological dynamics in pathogen populations. Although the field has witnessed a rich development of statistical inference tools with increasing levels of sophistication, these tools initially focused on sequences as their sole primary data source. Integrating various sources of information, however, promises to deliver more precise insights in infectious diseases and to increase opportunities for statistical hypothesis testing. Here, we review how the emerging concept of data integration is stimulating new advances in Bayesian evolutionary inference methodology which formalize a marriage of statistical thinking and evolutionary biology. These approaches include connecting sequence to trait evolution, such as for host, phenotypic and geographic sampling information, but also the incorporation of covariates of evolutionary and epidemic processes in the reconstruction procedures. We highlight how a full Bayesian approach to covariate modeling and testing can generate further insights into sequence evolution, trait evolution, and population dynamics in pathogen populations. Specific examples demonstrate how such approaches can be used to test the impact of host on rabies and HIV evolutionary rates, to identify the drivers of influenza dispersal as well as the determinants of rabies cross-species transmissions, and to quantify the evolutionary dynamics of influenza antigenicity. Finally, we briefly discuss how data integration is now also permeating through the inference of transmission dynamics, leading to novel insights into tree-generative processes and detailed reconstructions of transmission trees. [Bayesian inference; birth–death models; coalescent models; continuous trait evolution; covariates; data integration; discrete trait evolution; pathogen phylodynamics.
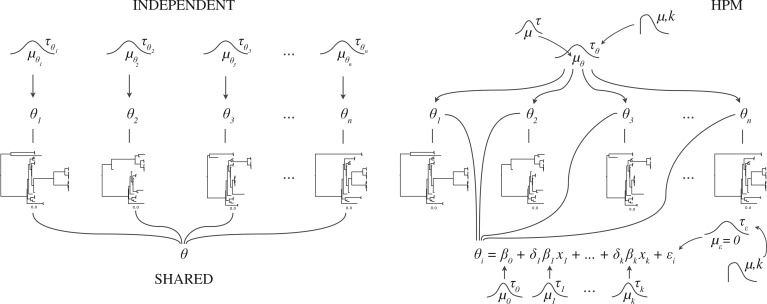
Figures




References
-
- Alizon S., von Wyl V., Stadler T., Kouyos R.D., Yerly S., Hirschel B., Boni J., Shah C., Klimkait T., Furrer H., Rauch A., Vernazza P.L., Bernasconi E., Battegay M., Bürgisser P., Telenti A., Günthard H.F., Bonhoeffer S. the Swiss Cohort Study. 2. Phylogenetic approach reveals that virus genotype largely determines HIV set-point viral load. PLoS Path. 6:e1001123. - PMC - PubMed
-
- Ayres D.L., Darling A., Zwickl D.J., Beerli P., Holder M.T., Lewis P.O., Huelsenbeck J.P., Ronquist F., Swofford D.L., Cummings M.P., Rambaut A., Suchard M.A. 2. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 61:170–173. - PMC - PubMed
-
- Baele G., Lemey P. 2. Bayesian evolutionary model testing in the phylogenomics era: matching model complexity with computational efficiency. Bioinformatics 29:1970–1979. - PubMed
-
- Bahl J., Nelson M.I., Chan K.H., Chen R., Vijaykrishna D., Halpin R.A., Stockwell T.B., Lin X., Wentworth D.E., Ghedin E., Guan Y., Peiris J.S.M, Riley S., Rambaut A., Holmes E.C., Smith G.J.D. 2. Temporally structured metapopulation dynamics and persistence of influenza A H3N2 virus in humans. Proc. Natl. Acad. Sci. USA 108:19359–19364. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
