

Enhanced dependency of KRAS-mutant colorectal cancer cells on RAD51-dependent homologous recombination repair identified from genetic interactions in Saccharomyces cerevisiae
- PMID: 28173629
- PMCID: PMC5527460
- DOI: 10.1002/1878-0261.12040
Enhanced dependency of KRAS-mutant colorectal cancer cells on RAD51-dependent homologous recombination repair identified from genetic interactions in Saccharomyces cerevisiae
Abstract
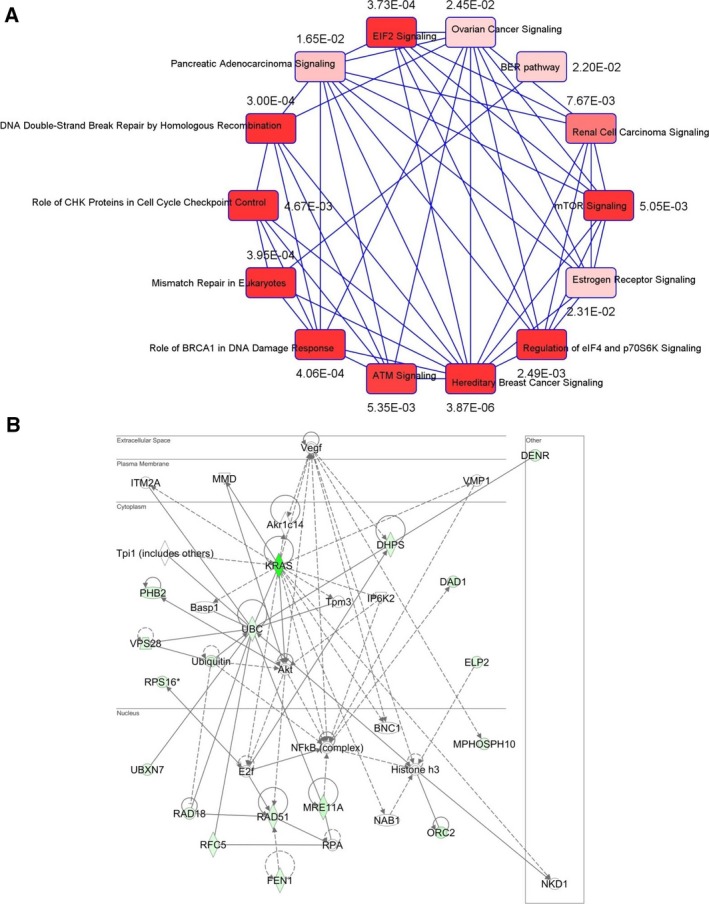
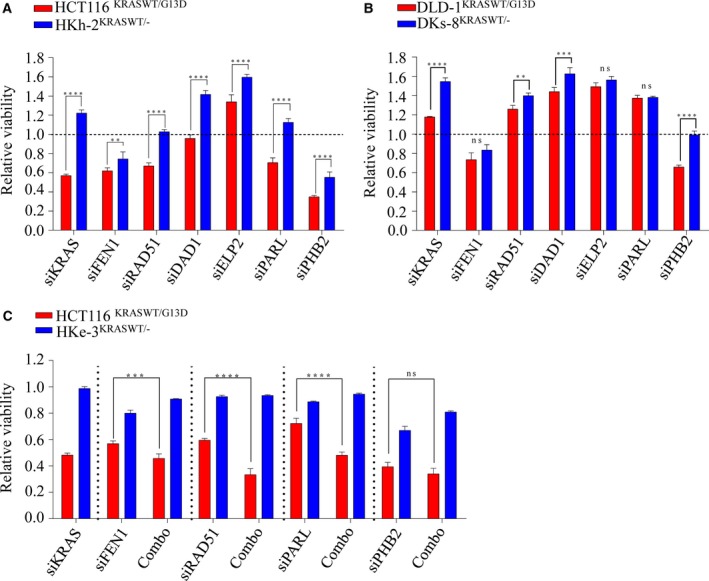
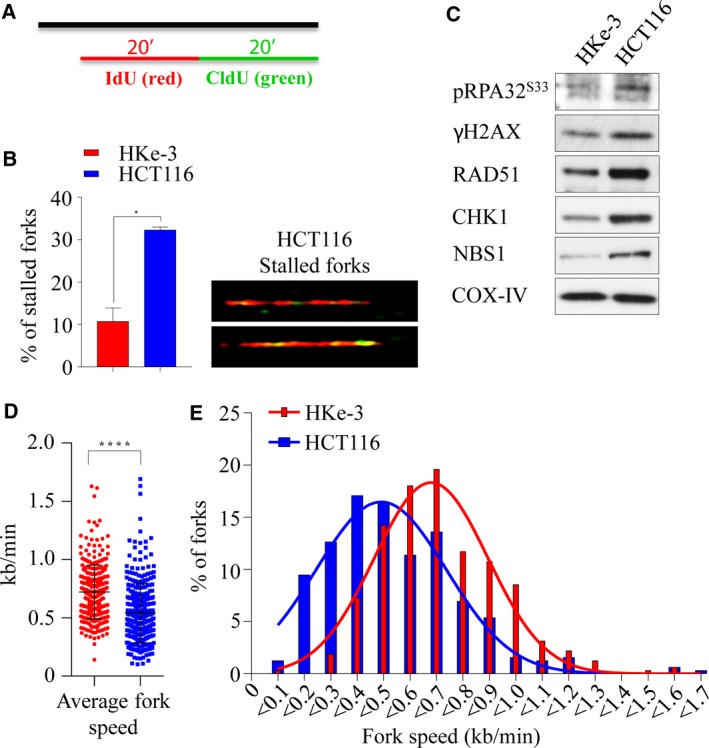
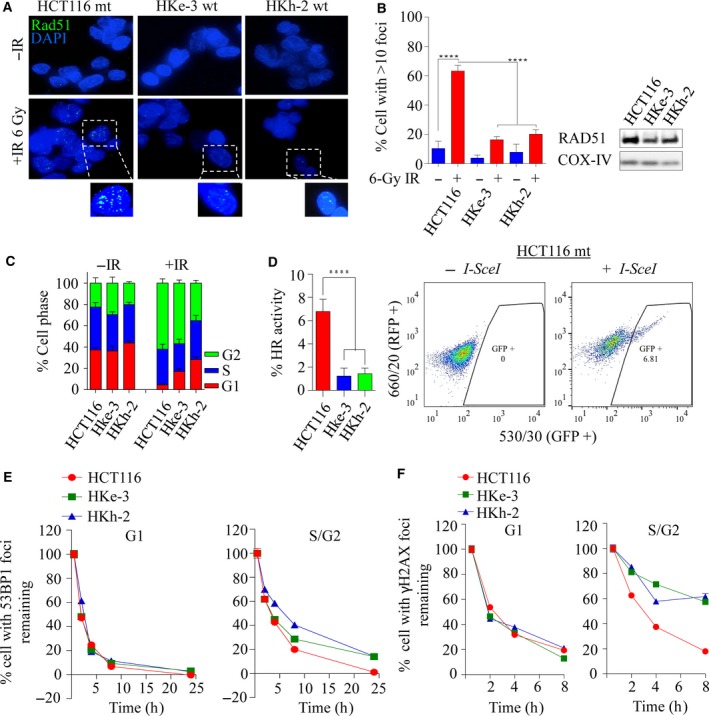
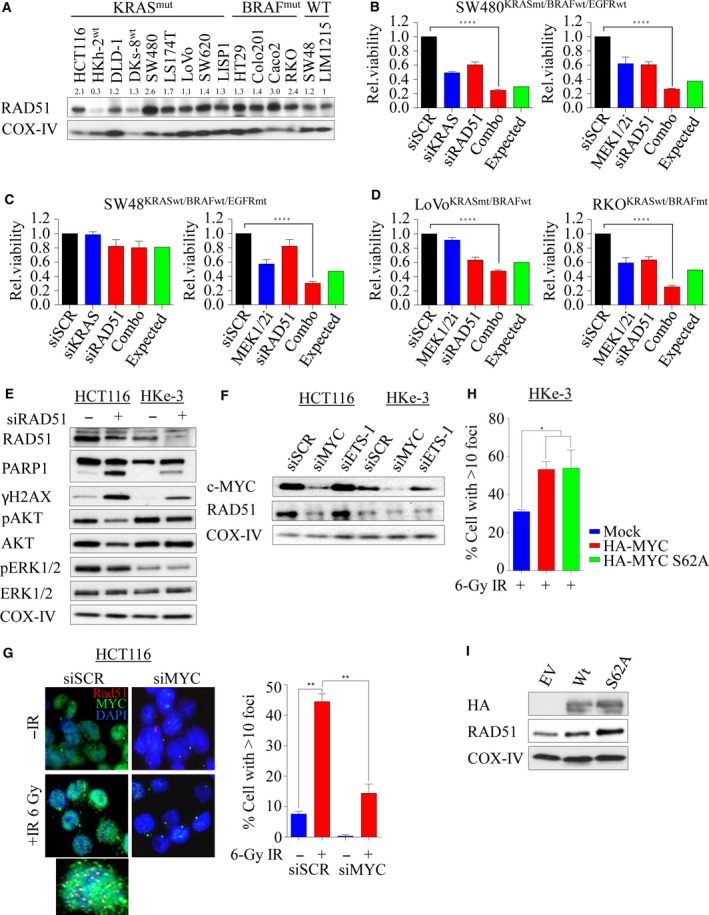
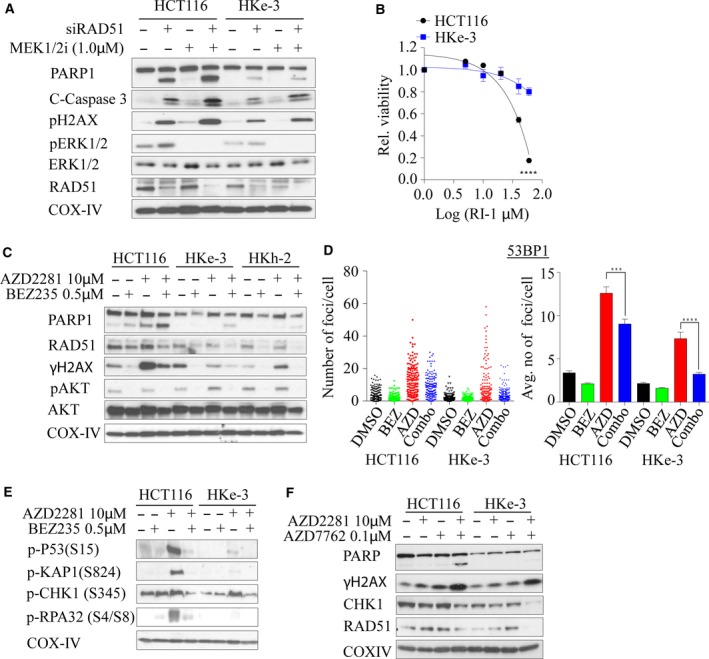
Activating KRAS mutations drive colorectal cancer tumorigenesis and influence response to anti-EGFR-targeted therapy. Despite recent advances in understanding Ras signaling biology and the revolution in therapies for melanoma using BRAF inhibitors, no targeted agents have been effective in KRAS-mutant cancers, mainly due to activation of compensatory pathways. Here, by leveraging the largest synthetic lethal genetic interactome in yeast, we identify that KRAS-mutated colorectal cancer cells have augmented homologous recombination repair (HRR) signaling. We found that KRAS mutation resulted in slowing and stalling of the replication fork and accumulation of DNA damage. Moreover, we found that KRAS-mutant HCT116 cells have an increase in MYC-mediated RAD51 expression with a corresponding increase in RAD51 recruitment to irradiation-induced DNA double-strand breaks (DSBs) compared to genetically complemented isogenic cells. MYC depletion using RNA interference significantly reduced IR-induced RAD51 foci formation and HRR. On the contrary, overexpression of either HA-tagged wild-type (WT) MYC or phospho-mutant S62A increased RAD51 protein levels and hence IR-induced RAD51 foci. Likewise, depletion of RAD51 selectively induced apoptosis in HCT116-mutant cells by increasing DSBs. Pharmacological inhibition targeting HRR signaling combined with PARP inhibition selectivity killed KRAS-mutant cells. Interestingly, these differences were not seen in a second isogenic pair of KRAS WT and mutant cells (DLD-1), likely due to their nondependency on the KRAS mutation for survival. Our data thus highlight a possible mechanism by which KRAS-mutant-dependent cells drive HRR in vitro by upregulating MYC-RAD51 expression. These data may offer a promising therapeutic vulnerability in colorectal cancer cells harboring otherwise nondruggable KRAS mutations, which warrants further investigation in vivo.
Keywords: KRAS; RAD51; DNA damage response; colorectal cancer; homologous recombination repair; therapeutic vulnerability.
© 2017 The Authors. Published by FEBS Press and John Wiley & Sons Ltd.
Figures
References
-
- Al‐Ejeh F, Simpson PT, Sanus JM, Klein K, Kalimutho M, Shi W, Miranda M, Kutasovic J, Raghavendra A, Madore J et al (2014) Meta‐analysis of the global gene expression profile of triple‐negative breast cancer identifies genes for the prognostication and treatment of aggressive breast cancer. Oncogenesis 3, e100. - PMC - PubMed
-
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC et al (2006) Oncogene‐induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637. - PubMed
-
- Baumann P and West SC (1998) Role of the human RAD51 protein in homologous recombination and double‐stranded‐break repair. Trends Biochem Sci 23, 247–251. - PubMed
-
- Bos JL, Rehmann H and Wittinghofer A (2007) GEFs and GAPs: critical elements in the control of small G proteins. Cell 129, 865–877. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
