

SGK3 sustains ERα signaling and drives acquired aromatase inhibitor resistance through maintaining endoplasmic reticulum homeostasis
- PMID: 28174265
- PMCID: PMC5338386
- DOI: 10.1073/pnas.1612991114
SGK3 sustains ERα signaling and drives acquired aromatase inhibitor resistance through maintaining endoplasmic reticulum homeostasis
Abstract
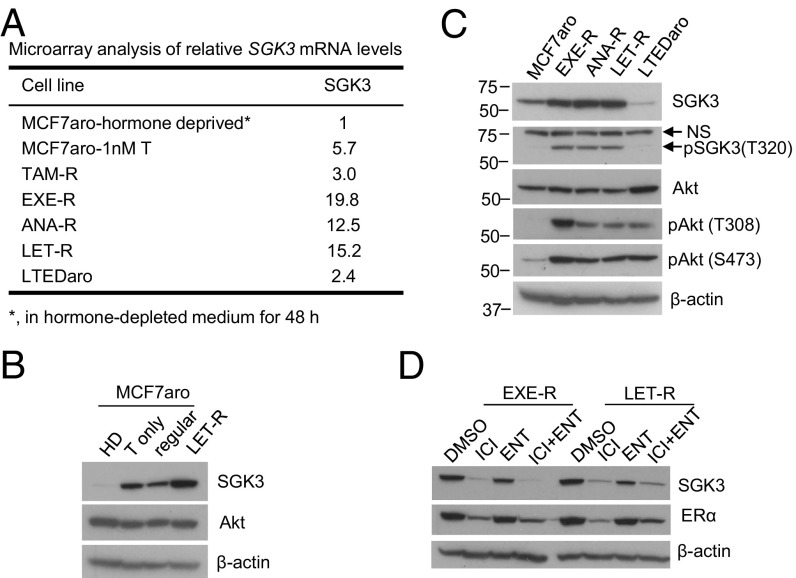
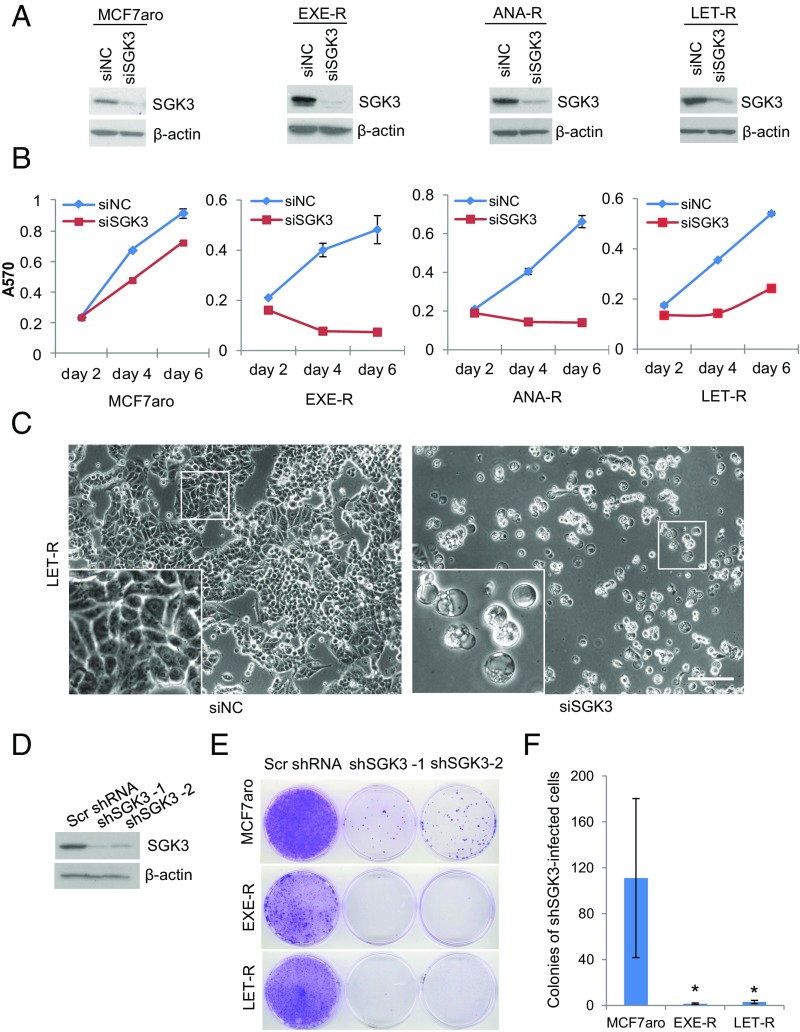
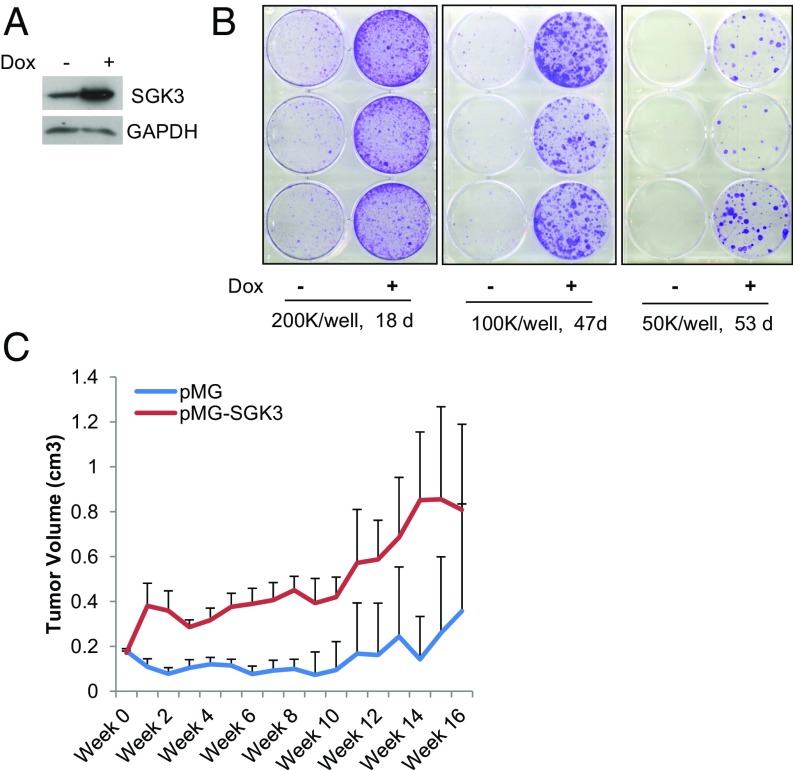
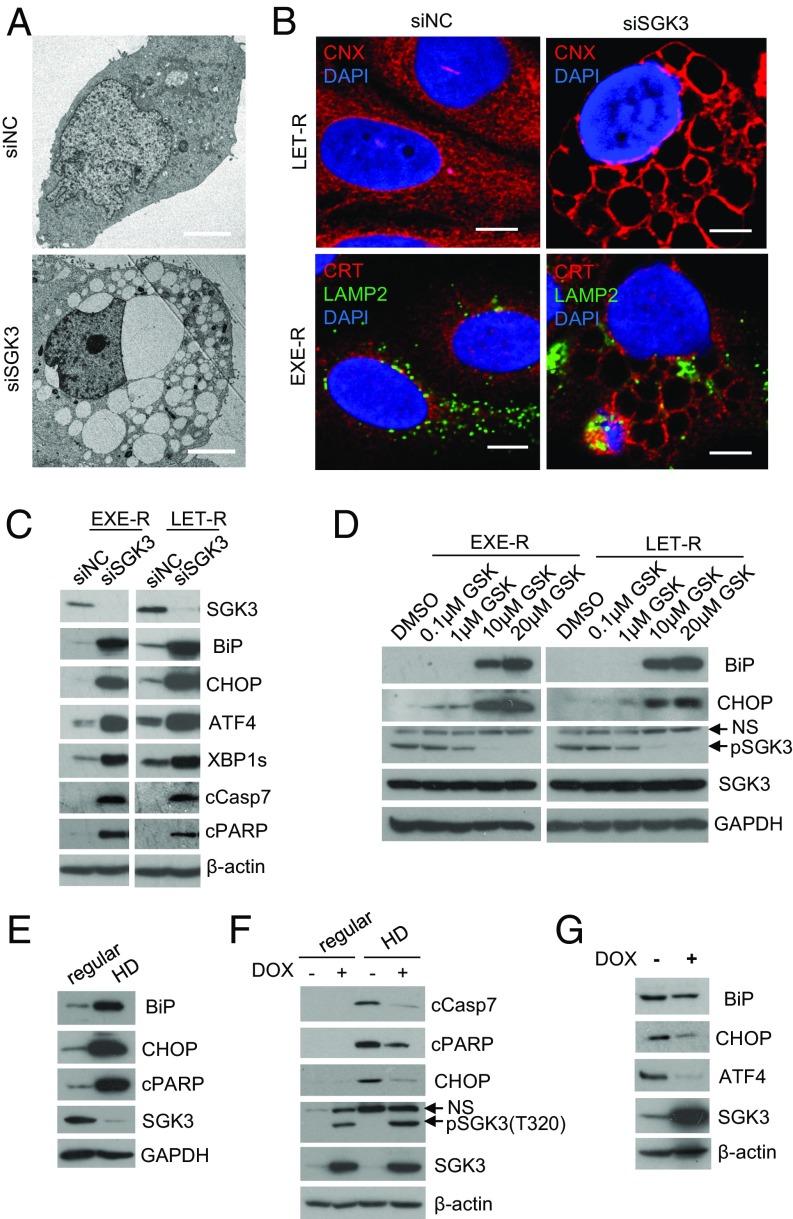
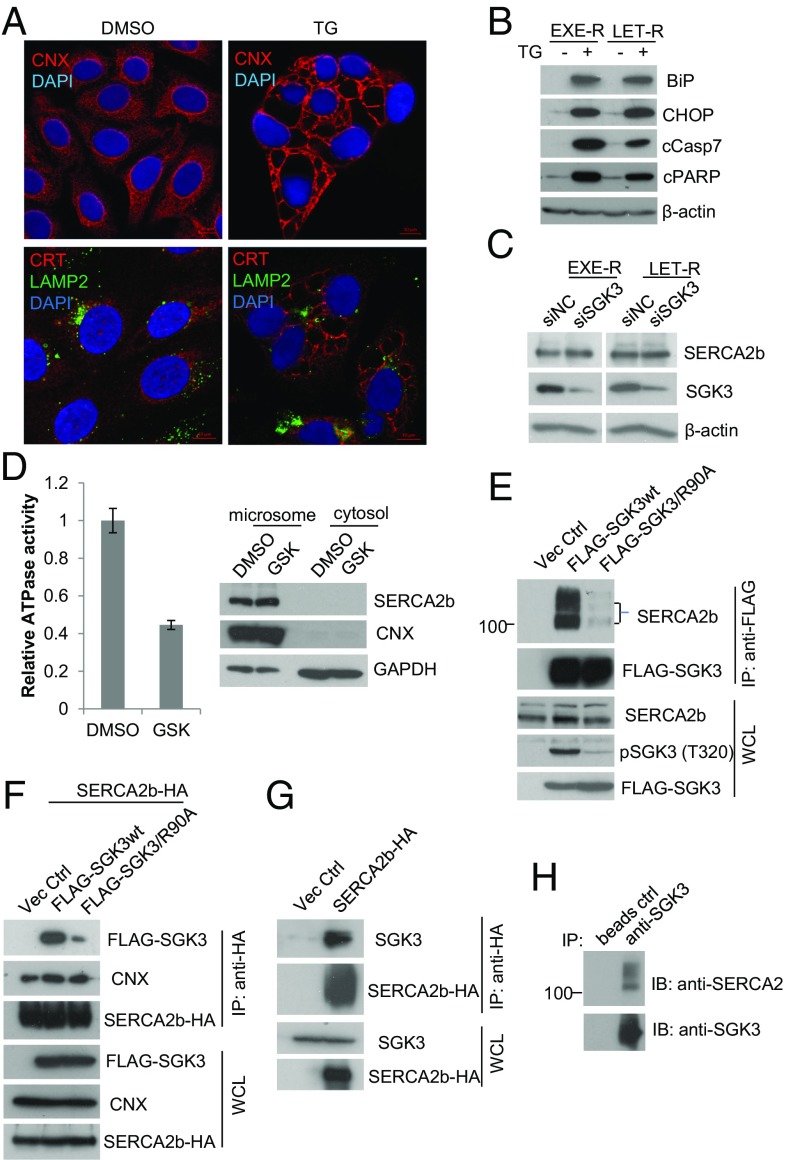
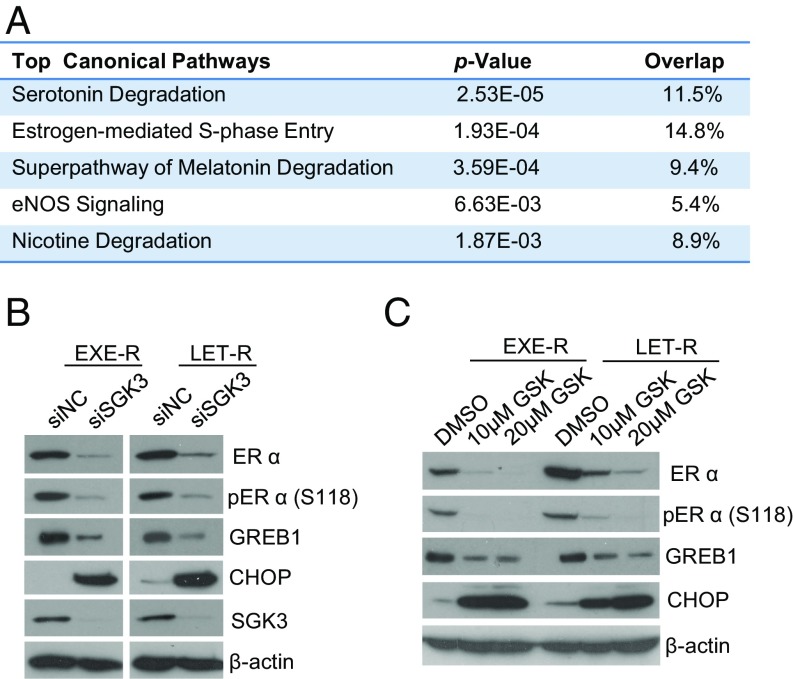
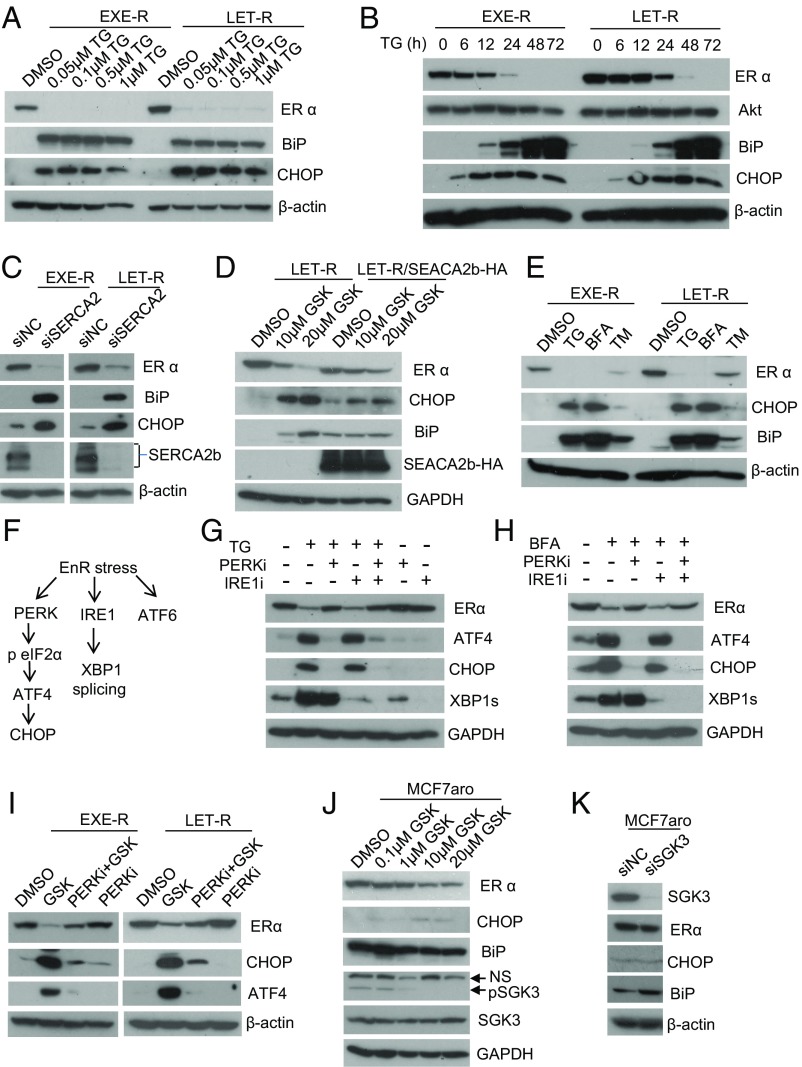
Many estrogen receptor alpha (ERα)-positive breast cancers initially respond to aromatase inhibitors (AIs), but eventually acquire resistance. Here, we report that serum- and glucocorticoid-inducible kinase 3 (SGK3), a kinase transcriptionally regulated by ERα in breast cancer, sustains ERα signaling and drives acquired AI resistance. SGK3 is up-regulated and essential for endoplasmic reticulum (EnR) homeostasis through preserving sarcoplasmic/EnR calcium ATPase 2b (SERCA2b) function in AI-resistant cells. We have further found that EnR stress response down-regulates ERα expression through the protein kinase RNA-like EnR kinase (PERK) arm, and SGK3 retains ERα expression and signaling by preventing excessive EnR stress. Our study reveals regulation of ERα expression mediated by the EnR stress response and the feed-forward regulation between SGK3 and ERα in breast cancer. Given SGK3 inhibition reduces AI-resistant cell survival by eliciting excessive EnR stress and also depletes ERα expression/function, we propose SGK3 inhibition as a potential effective treatment of acquired AI-resistant breast cancer.
Keywords: SERCA2; SGK3; aromatase inhibitor; endoplasmic reticulum stress; estrogen receptor.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Amphiregulin retains ERα expression in acquired aromatase inhibitor resistant breast cancer cells.Endocr Relat Cancer. 2020 Dec;27(12):671-683. doi: 10.1530/ERC-20-0258. Endocr Relat Cancer. 2020. PMID: 33112819 Free PMC article.
-
Expression of the K303R estrogen receptor-alpha breast cancer mutation induces resistance to an aromatase inhibitor via addiction to the PI3K/Akt kinase pathway.Cancer Res. 2009 Jun 1;69(11):4724-32. doi: 10.1158/0008-5472.CAN-08-4194. Cancer Res. 2009. PMID: 19487288 Free PMC article.
-
Expression of estrogen-related gene markers in breast cancer tissue predicts aromatase inhibitor responsiveness.PLoS One. 2013 Nov 6;8(11):e77543. doi: 10.1371/journal.pone.0077543. eCollection 2013. PLoS One. 2013. PMID: 24223121 Free PMC article.
-
An "omics" approach to determine the mechanisms of acquired aromatase inhibitor resistance.OMICS. 2011 Jun;15(6):347-52. doi: 10.1089/omi.2010.0097. Epub 2011 Feb 19. OMICS. 2011. PMID: 21332390 Free PMC article. Review.
-
Identification of miRNAs as biomarkers for acquired endocrine resistance in breast cancer.Mol Cell Endocrinol. 2017 Nov 15;456:76-86. doi: 10.1016/j.mce.2017.02.004. Epub 2017 Feb 3. Mol Cell Endocrinol. 2017. PMID: 28163101 Review.
Cited by
-
Ionizing radiation downregulates estradiol synthesis via endoplasmic reticulum stress and inhibits the proliferation of estrogen receptor-positive breast cancer cells.Cell Death Dis. 2021 Oct 29;12(11):1029. doi: 10.1038/s41419-021-04328-w. Cell Death Dis. 2021. PMID: 34716300 Free PMC article.
-
Potential implication of SGK1-dependent activity change in BV-2 microglial cells.Int J Physiol Pathophysiol Pharmacol. 2018 Apr 20;10(2):115-123. eCollection 2018. Int J Physiol Pathophysiol Pharmacol. 2018. PMID: 29755644 Free PMC article.
-
AKTIP loss is enriched in ERα-positive breast cancer for tumorigenesis and confers endocrine resistance.Cell Rep. 2022 Dec 13;41(11):111821. doi: 10.1016/j.celrep.2022.111821. Cell Rep. 2022. PMID: 36516775 Free PMC article.
-
Maresin 1 attenuates NAFLD by suppression of endoplasmic reticulum stress via AMPK-SERCA2b pathway.J Biol Chem. 2018 Mar 16;293(11):3981-3988. doi: 10.1074/jbc.RA117.000885. Epub 2018 Feb 5. J Biol Chem. 2018. PMID: 29414781 Free PMC article.
-
The SGK3/GSK3β/β-catenin signaling promotes breast cancer stemness and confers resistance to alpelisib therapy.Int J Biol Sci. 2025 Mar 19;21(6):2462-2475. doi: 10.7150/ijbs.104850. eCollection 2025. Int J Biol Sci. 2025. PMID: 40303291 Free PMC article.
References
-
- Ma CX, Reinert T, Chmielewska I, Ellis MJ. Mechanisms of aromatase inhibitor resistance. Nat Rev Cancer. 2015;15(5):261–275. - PubMed
-
- Robertson JF, et al. Endocrine treatment options for advanced breast cancer--the role of fulvestrant. Eur J Cancer. 2005;41(3):346–356. - PubMed
-
- Ali S, Coombes RC. Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002;2(2):101–112. - PubMed
-
- Krebs J, Agellon LB, Michalak M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem Biophys Res Commun. 2015;460(1):114–121. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
