

RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome
- PMID: 28176780
- PMCID: PMC5309790
- DOI: 10.1038/ncomms14329
RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome
Abstract
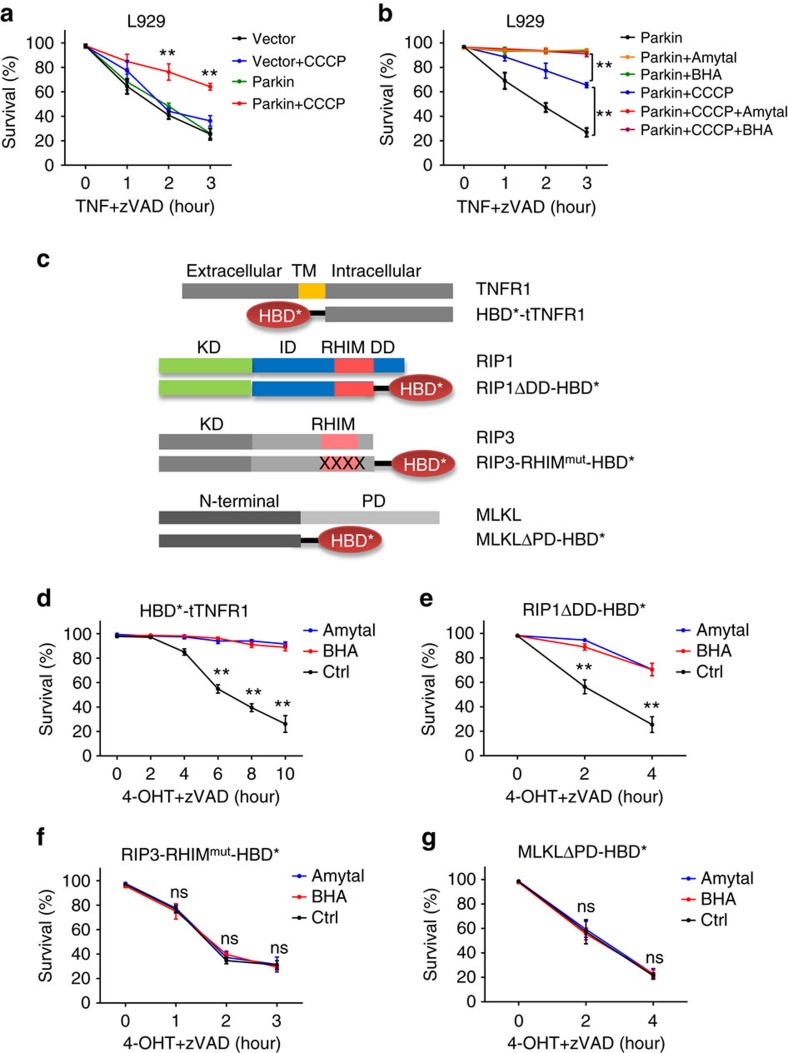
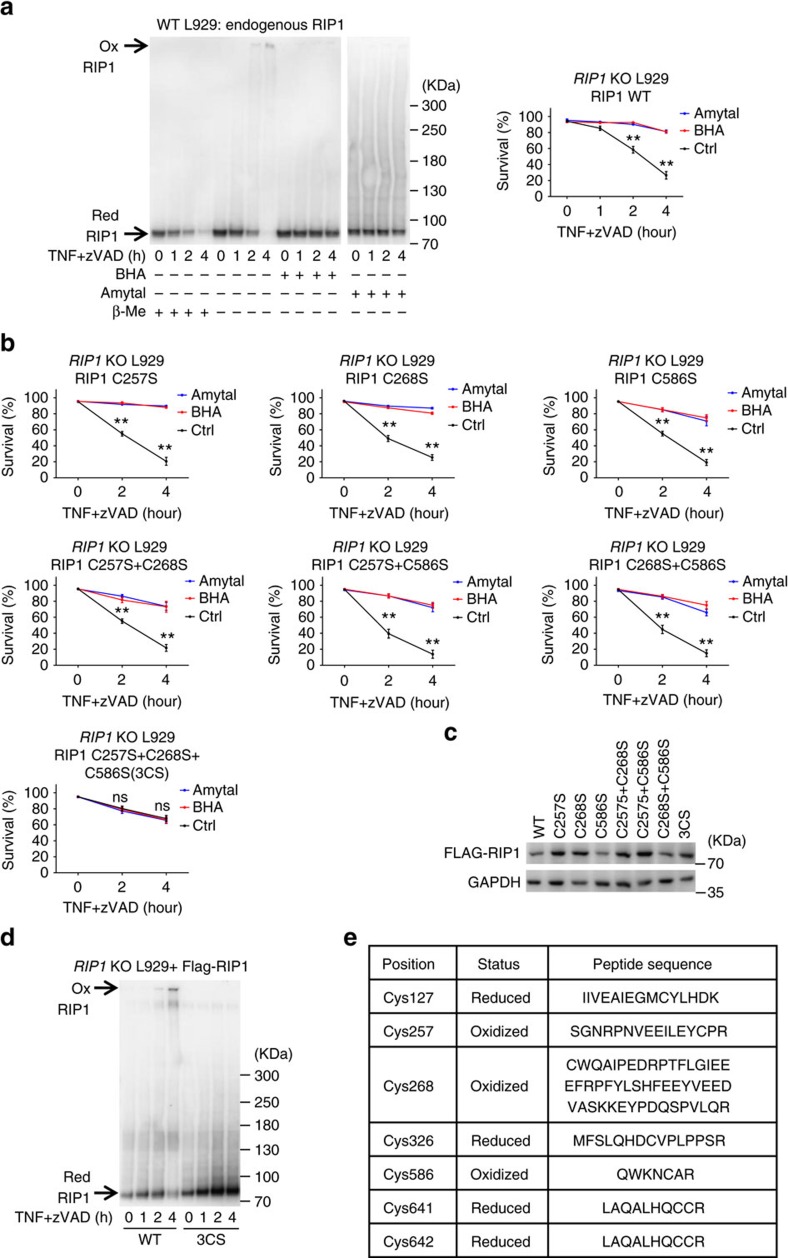
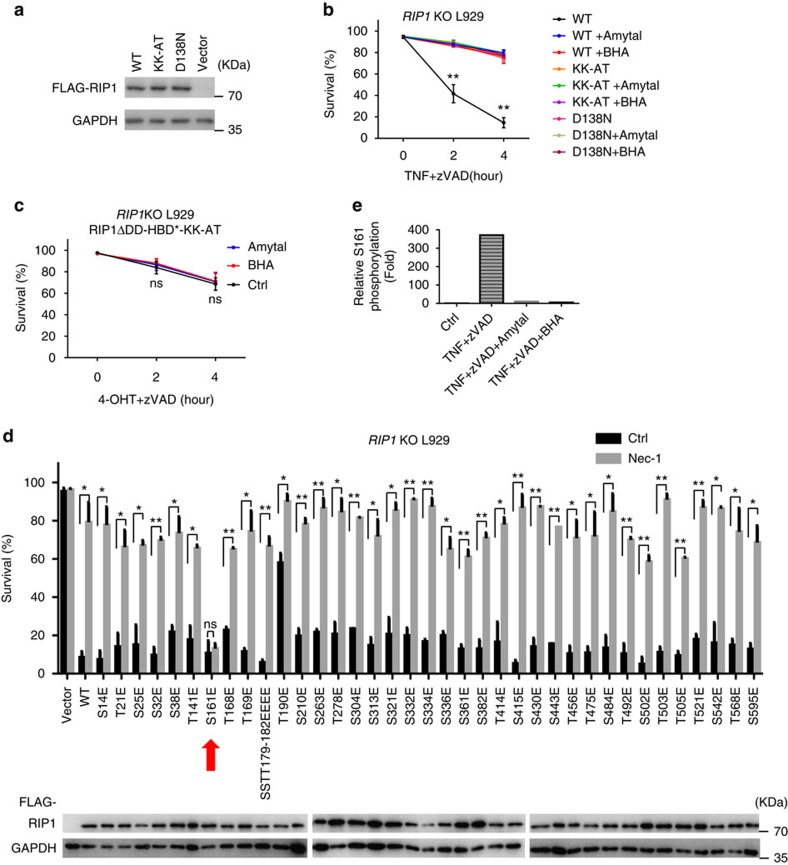
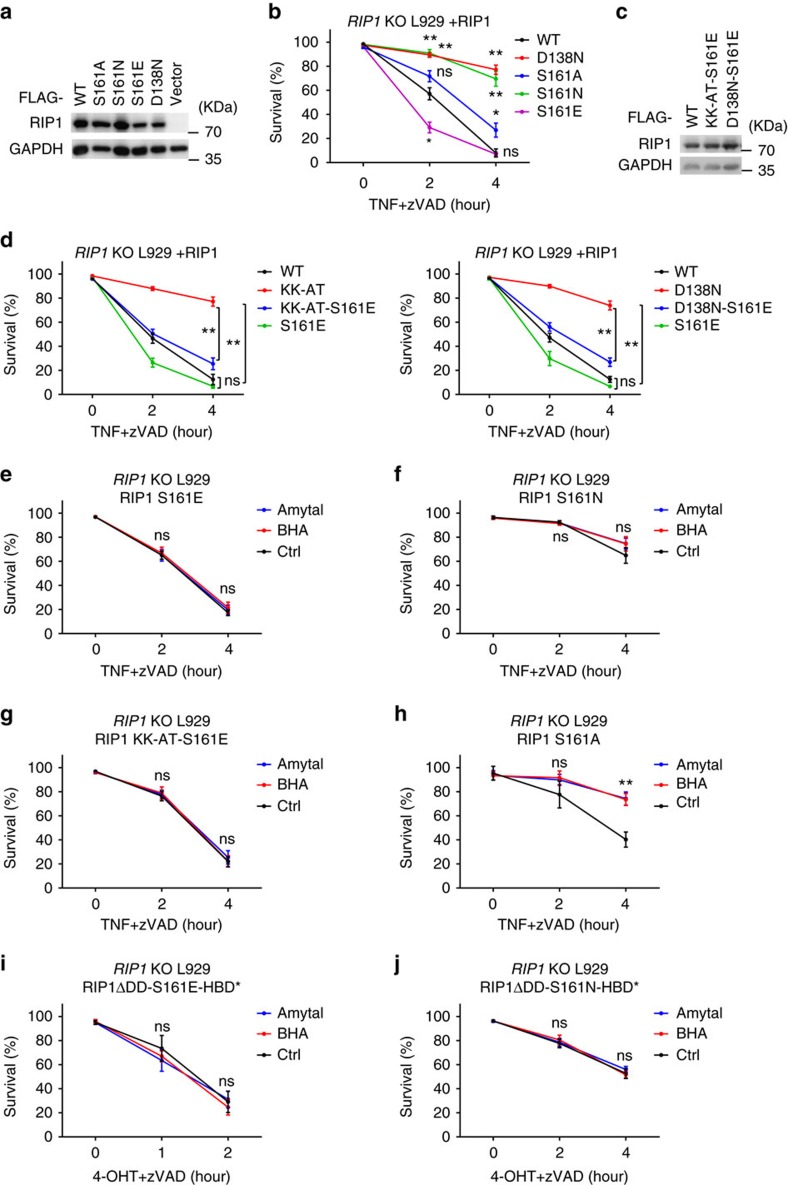
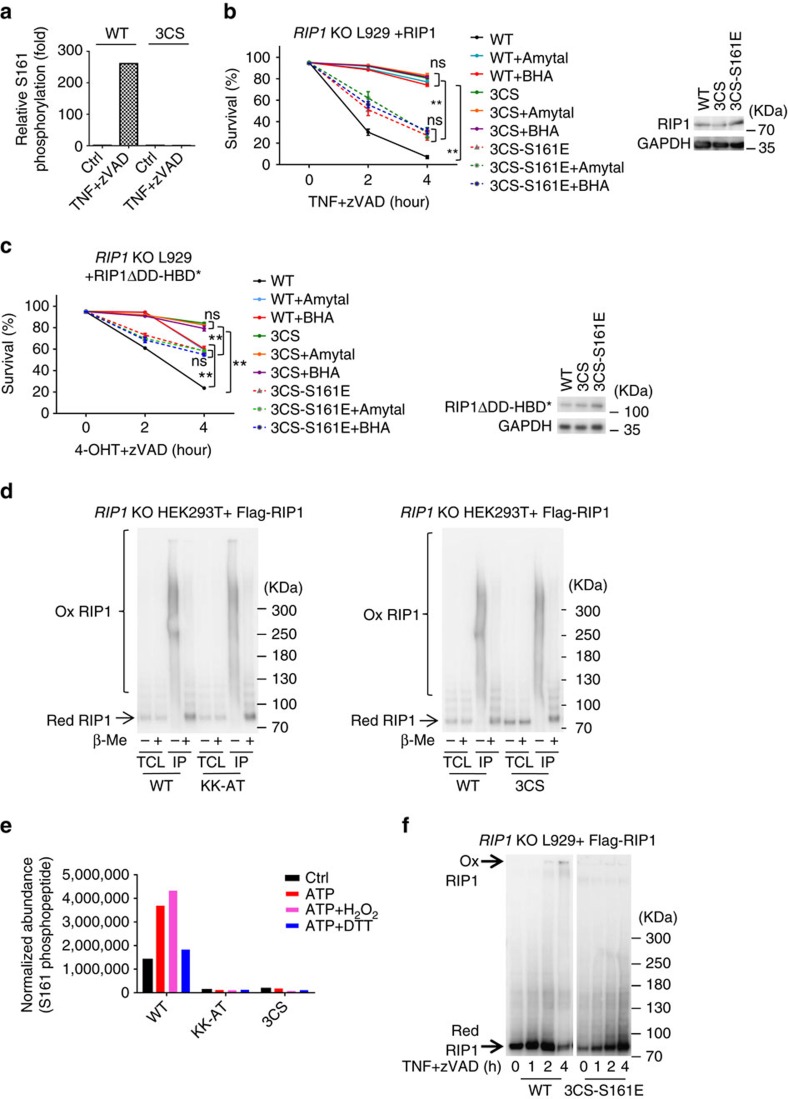
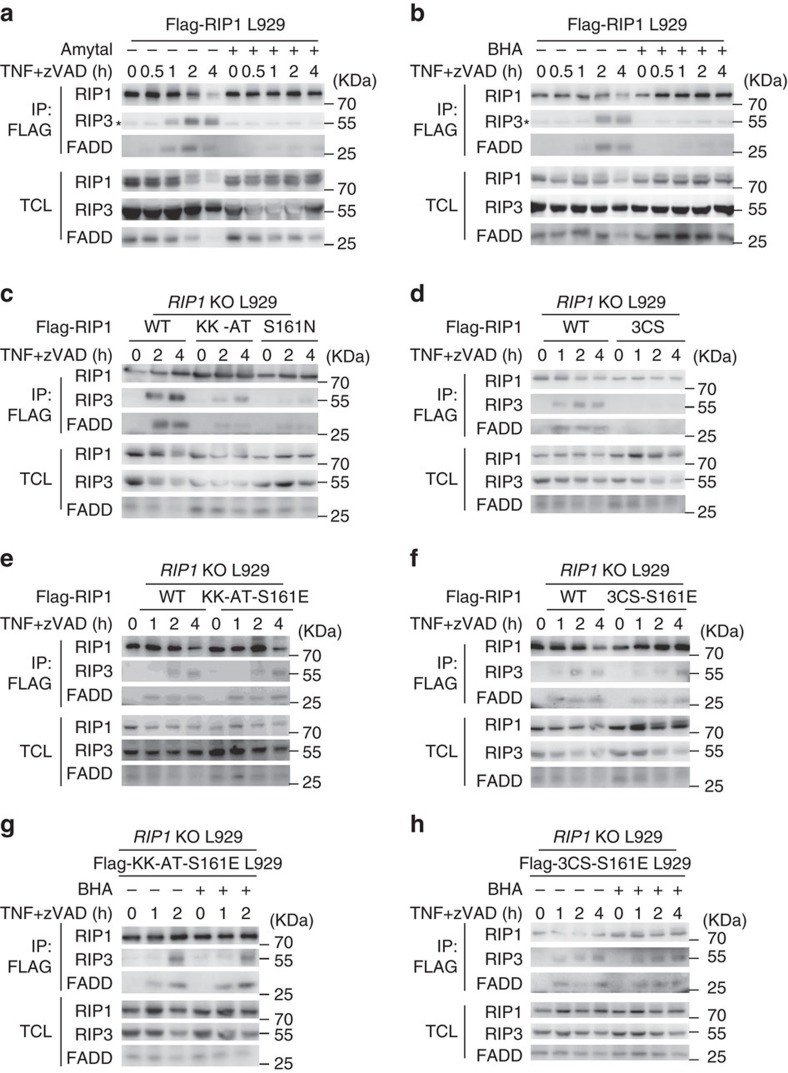
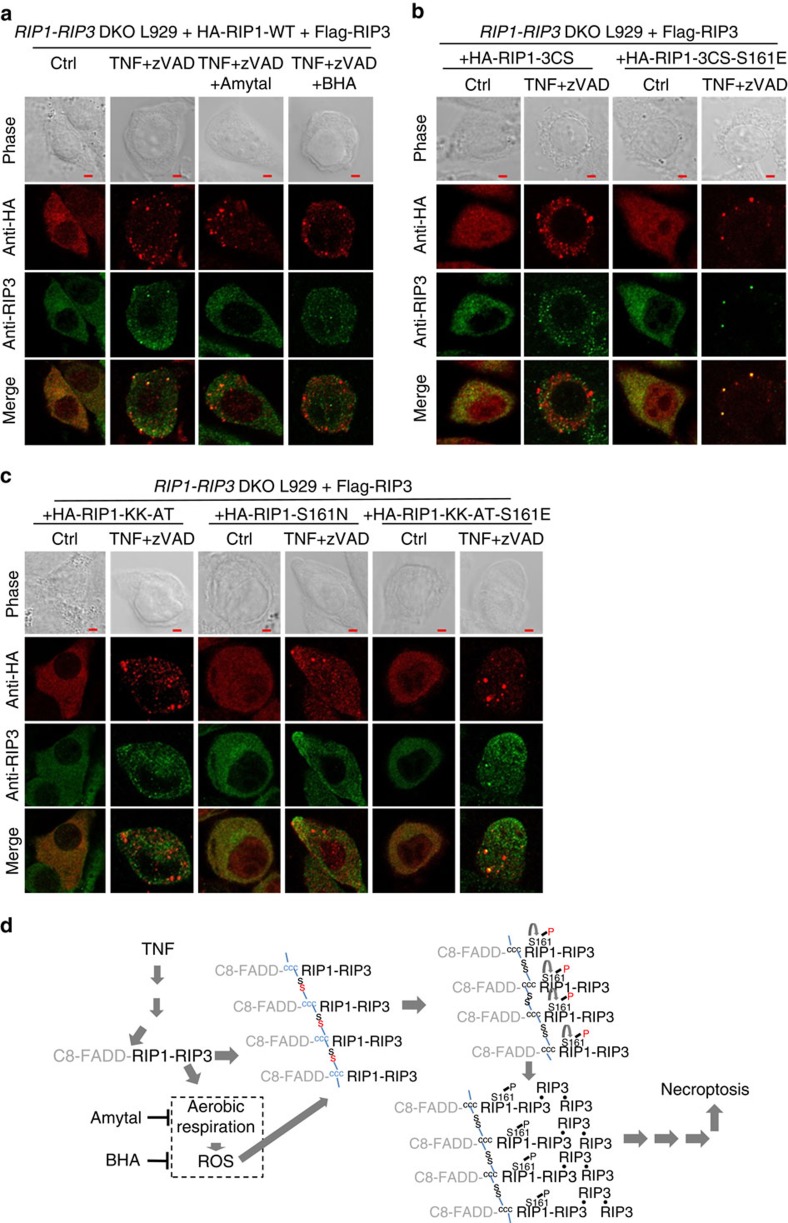
Necroptosis is a type of programmed cell death with great significance in many pathological processes. Tumour necrosis factor-α(TNF), a proinflammatory cytokine, is a prototypic trigger of necroptosis. It is known that mitochondrial reactive oxygen species (ROS) promote necroptosis, and that kinase activity of receptor interacting protein 1 (RIP1) is required for TNF-induced necroptosis. However, how ROS function and what RIP1 phosphorylates to promote necroptosis are largely unknown. Here we show that three crucial cysteines in RIP1 are required for sensing ROS, and ROS subsequently activates RIP1 autophosphorylation on serine residue 161 (S161). The major function of RIP1 kinase activity in TNF-induced necroptosis is to autophosphorylate S161. This specific phosphorylation then enables RIP1 to recruit RIP3 and form a functional necrosome, a central controller of necroptosis. Since ROS induction is known to require necrosomal RIP3, ROS therefore function in a positive feedback circuit that ensures effective induction of necroptosis.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Christofferson D. E., Li Y. & Yuan J. Control of life-or-death decisions by RIP1 kinase. Annu. Rev. Physiol. 76, 129–150 (2014). - PubMed
-
- Han J., Zhong C. Q. & Zhang D. W. Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat. Immunol. 12, 1143–1149 (2011). - PubMed
-
- Sun L. & Wang X. A new kind of cell suicide: mechanisms and functions of programmed necrosis. Trends Biochem. Sci. 39, 587–593 (2014). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
