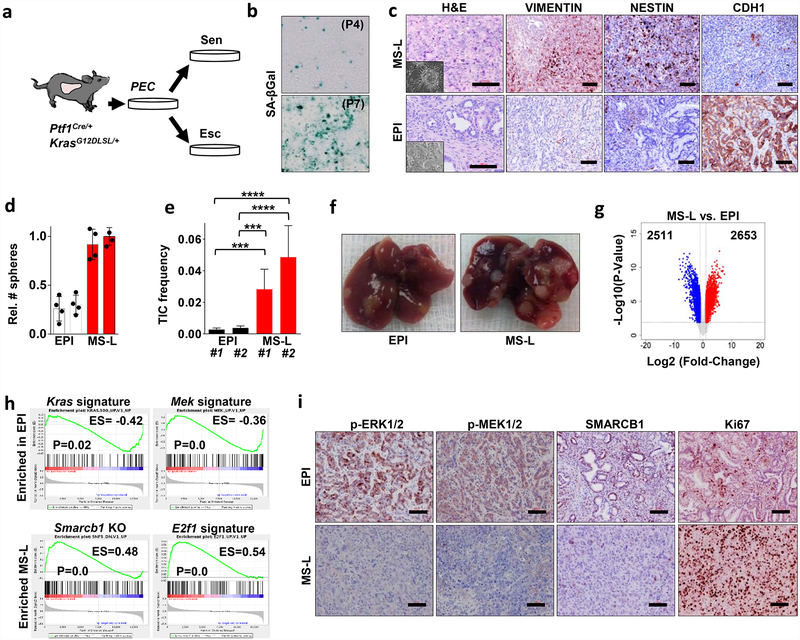
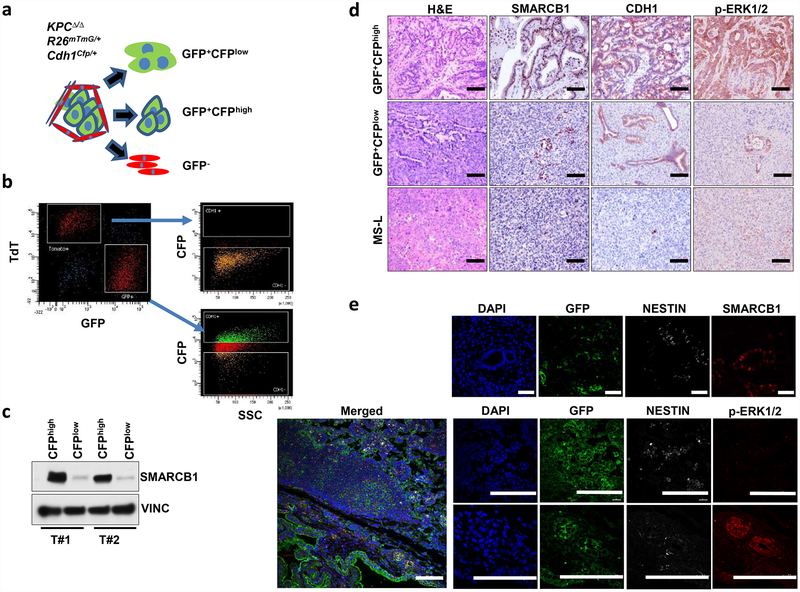
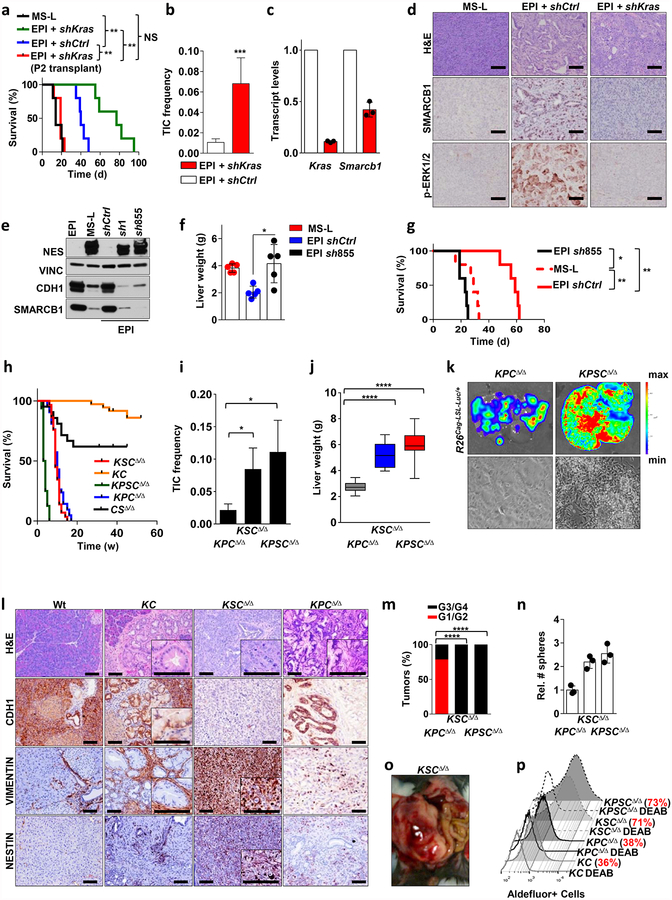
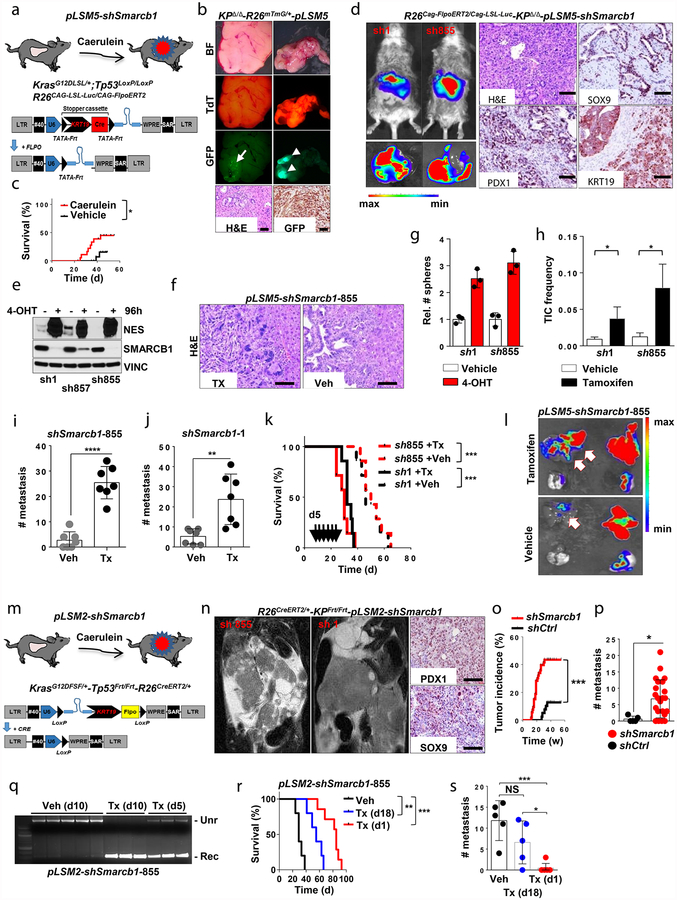
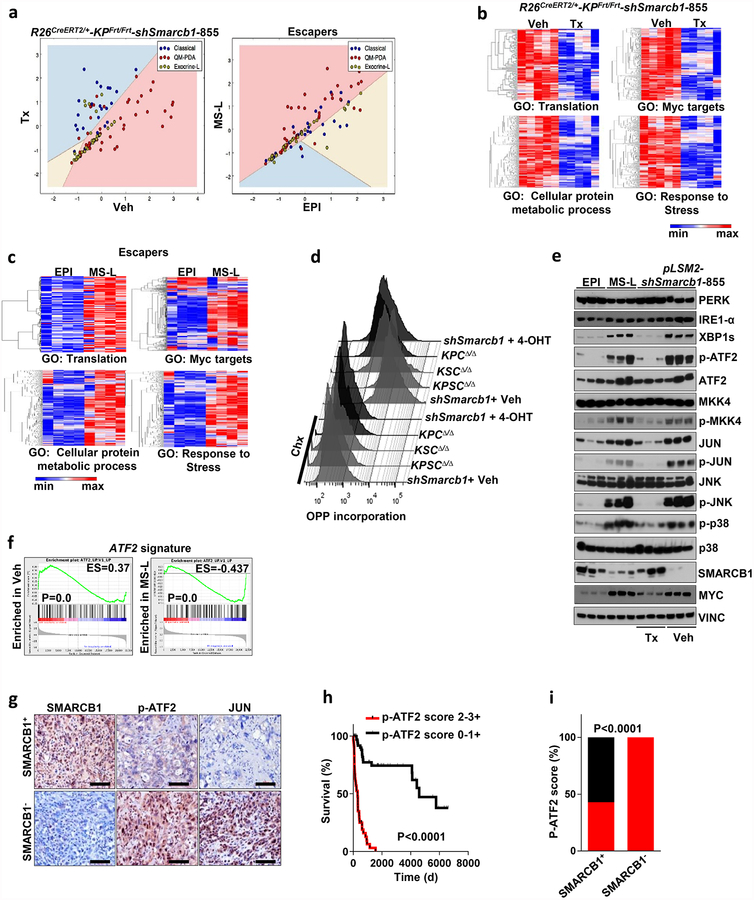
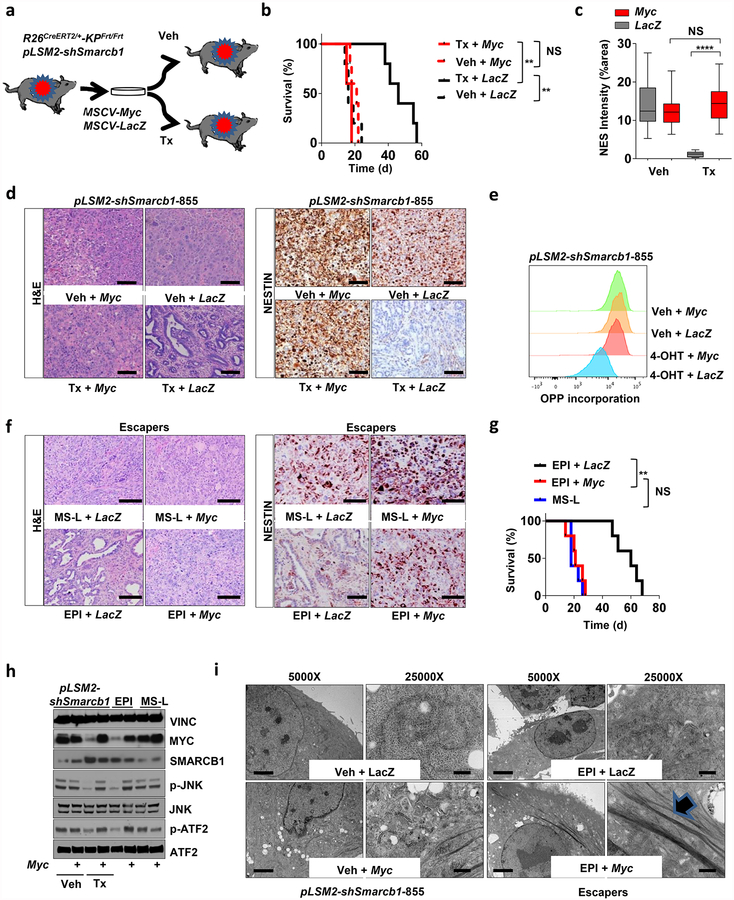
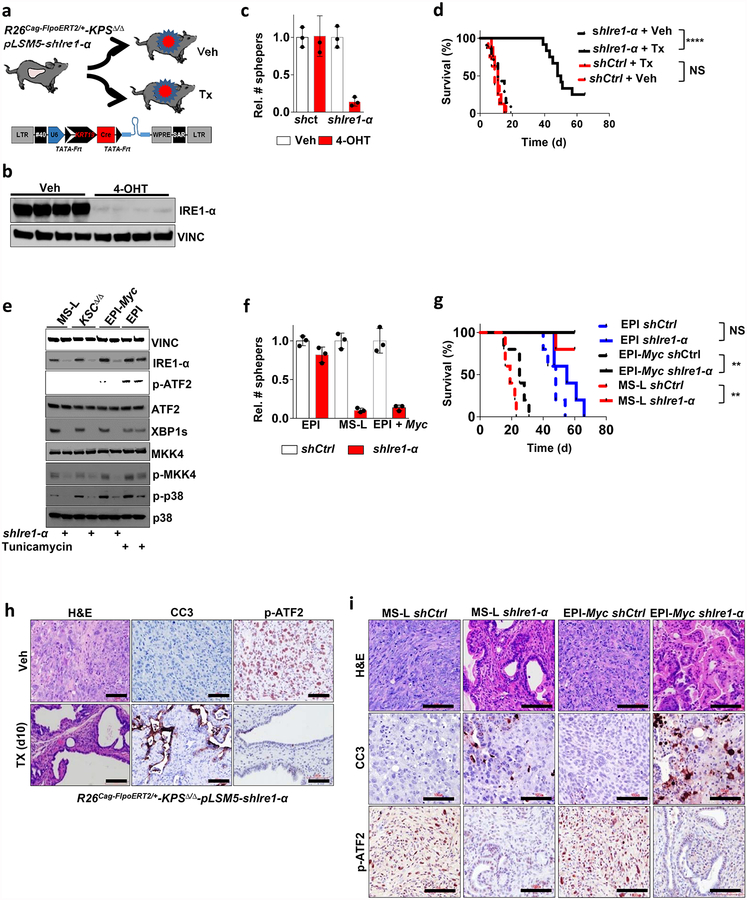
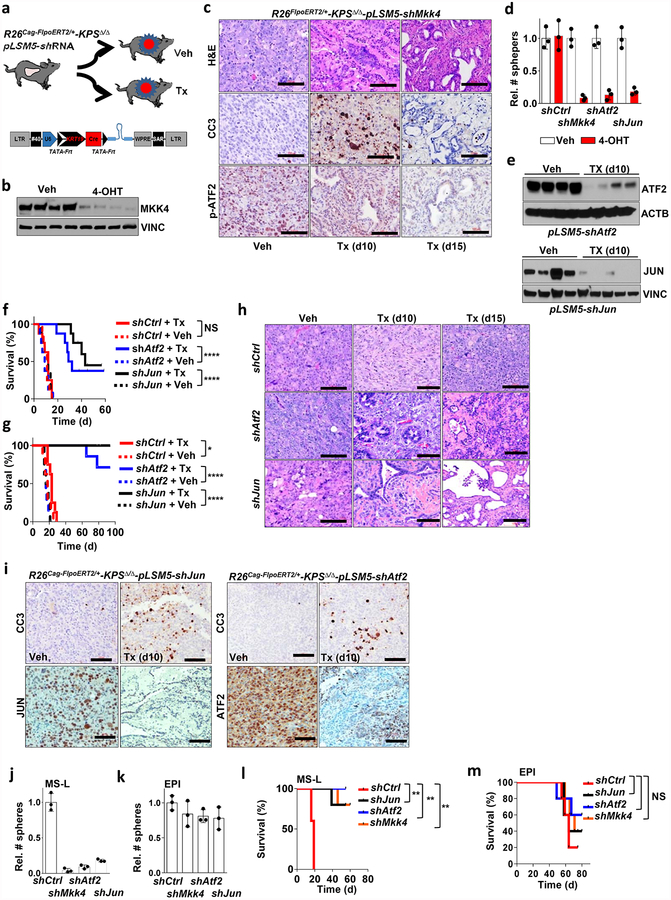
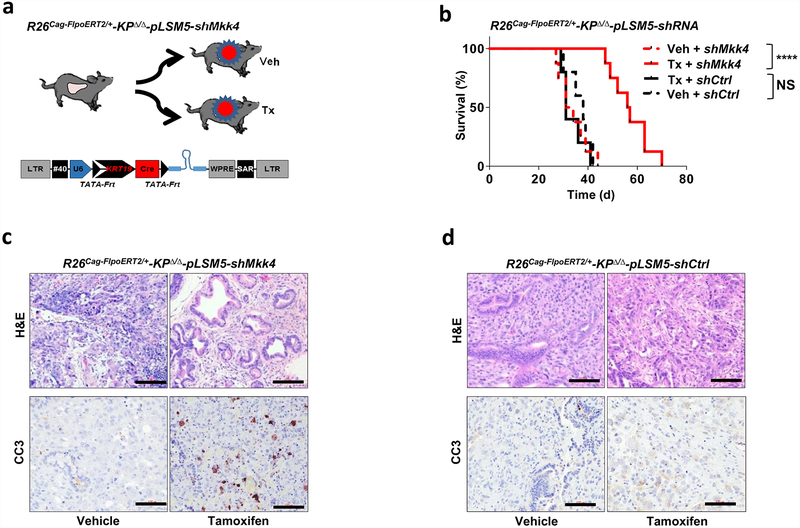
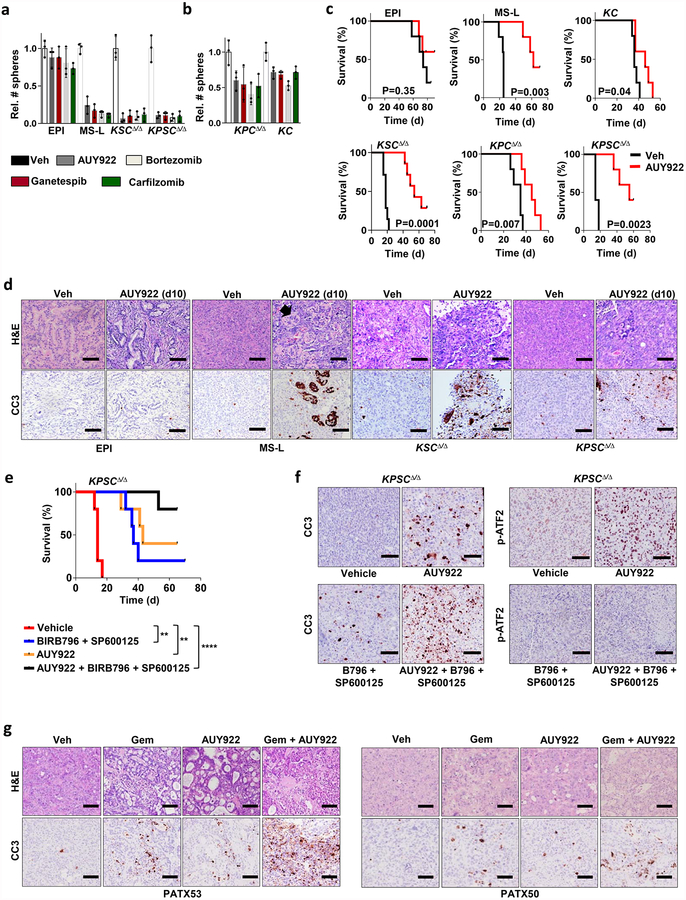
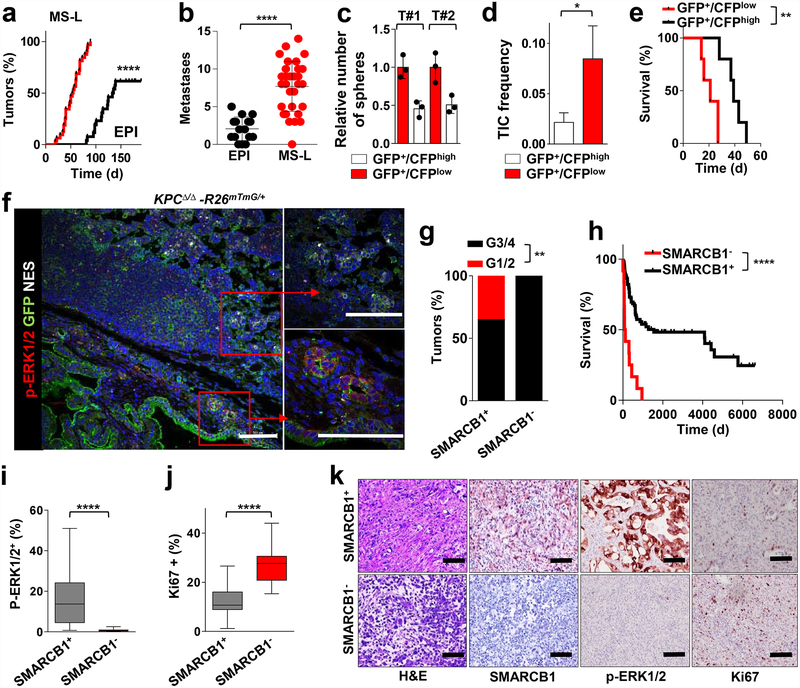
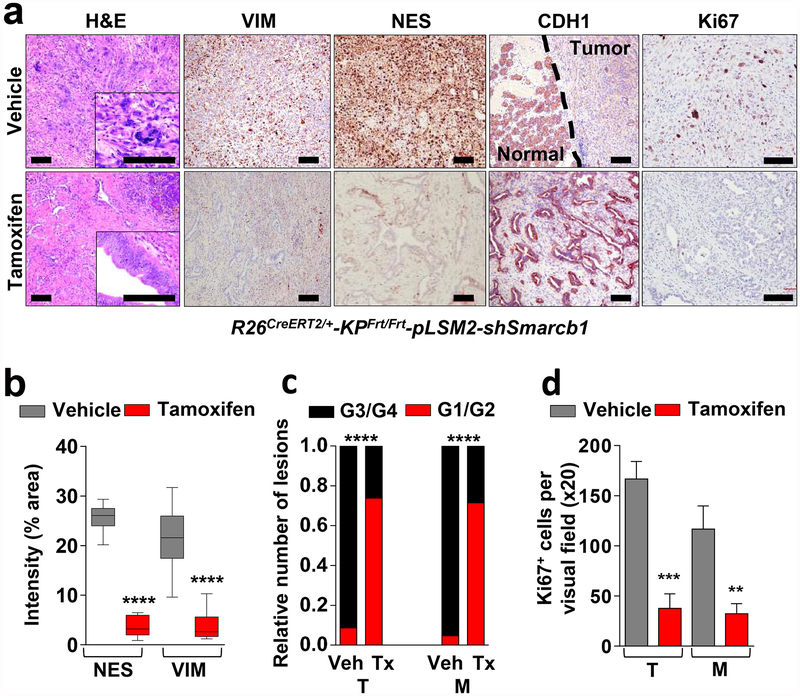
Extended Data Figure 4 |. Smarcb1 restrains the expansion of mesenchymal clones in PDAC.
a, Schematic showing the lentiviral construct for Flpo-mediated tissue-specific, time-restricted gene inactivation in vivo (pLSM5). High-titre-purified lentiviral particles (2–5 × 108 IU) were introduced surgically into the tail of the pancreas. At 7 days after surgery, mice were treated with caerulein to induce inflammation, proliferation and the expression of genes under the Krt19 promoter in the acinar compartment. The tissue specificity is provided by the human Krt19 promoter driving the expression of the Cre recombinase resulting in the activation of the latent mutant KrasG12DLSL/+, the inactivation of the conditional Tp53LoxP allele and the activation of the R26CAG-LSL-Luc reporter. The time-restricted activation of the shRNA is mediated by the ubiquitous R26Cag-FlpoERT2 inducible, codon-optimized Flpo recombinase upon tamoxifen treatment and removal of the Krt19-Cre stopper cassette, which is flanked by TATA-Frt sites. b, Representative panels showing the gross appearance of pancreata of KPΔ/Δ-R26mTmG/+ mice orthotopically injected with 108 viral particles, treated with caerulein 7 days after surgery and killed at the 3 weeks (left) or 3 months (right). Arrow indicates mosaic activation of the GFP reporter at 3 weeks. Arrowhead indicates GFP+ tumour nodules at 3 months. TdT, TdTomato. c, Kaplan–Meier analysis of tumour incidence in KPΔ/Δ-R26mTmG/+ mice orthotopically injected with 108 particles of the pLSM5-K19-Cre vector and assigned to caerulein and vehicle treatment. Caerulein treatment resulted in increased tumour incidence and decreased latency when compared to vehicle control (n = 18 per group). d, Representative luciferase images (left) and pathological characterization (right) of tumours generated with lentiviral-mosaic-somatic technology (R26Cag-FlpoERT2/Cag-LSL-Luc-KPΔ/Δ-pLSM5-shSmarcb1-1 or R26Cag-FlpoERT2/Cag-LSL-Luc-KPΔ/Δ-pLSM5-shSmarcb1-855). Orthotopic tumours harbouring latent Flpo/Frt-dependent, tamoxifen-inducible shRNAs against Smarcb1 were generated into a Kras-mutant, Tp53-deficient background (KPΔ/Δ). Mice were monitored weekly for tumour growth by bioluminescence imaging and lesions were characterized by immunophenotypic analysis for pancreatic lineage-differentiation markers before functional studies. e, Expression levels of SMARCB1 and nestin in short-term cultures established from tumours generated in R26Cag-FlpoERT2/Cag-LSL-Luc-KPΔ/Δ-pLSM5-shSmarcb1 mice assessed by western blot. Protein lysates were collected 96 h after 4-OHT (4-hydroxytamoxifen) treatment. Vinculin was used as loading control. f, Representative images of liver metastasis stained for haematoxylin and eosin from tamoxifen- and vehicle-treated R26Cag-FlpoERT2/Cag-LSL-Luc-KPΔ/Δ-pLSM5-shSmarcb1-855 mice. Tamoxifen-driven acute ablation of Smarcb1 in vivo results in poorly differentiated, invasive lesions. g, Spherogenic potential of low-passage spheroids obtained from R26Cag-FlpoERT2/Cag-LSL-Luc- KPΔ/Δ-pLSM5-shSmarcb1-1 and R26Cag-FlpoERT2/Cag-LSL-Luc-KPΔ/Δ-pLSM5-shSmarcb1-855 tumours upon treatment with 4-hydroxytamoxifen or vehicle control (n = 3 per group). Data are mean ± s.d. of technical replicates (one representative experiment out of three). The acute ablation of Smarcb1 results in a robust increase of the clonogenic potential in the KPΔ/Δ background. h, TIC frequency of KPΔ/Δ-R26Cag-FlpoERT2/Cag-LSL-Luc-pLSM5-shSmarcb1-1 and R26Cag-FlpoERT2/Cag-LSL-Luc-KPΔ/Δ-pLSM5-shSmarcb1-855 tumours upon treatment with vehicle or tamoxifen assessed by limiting-dilution experiments in immunocompromised (NCr Nude) mice (sh1 and vehicle, n = 25; sh1 and tamoxifen, n = 20; sh855 and vehicle, n = 19; sh855 and tamoxifen, n = 19) and calculated using the L-Calc software. Data are mean proportion of TIC ± s.e.m. i, j, Metastatic burden assesses by counting the number of superficial liver, peritoneal and lung metastases in NCr Nude mice transplanted orthotopically with R26Cag-FlpoERT2/Cag-LSL-Luc- KPΔ/Δ-pLSM5-shSmarcb1-1 or R26Cag-FlpoERT2/Cag-LSL-Luc-KPΔ/Δ-pLSM5-shSmarcb1-855 tumours assigned to vehicle or tamoxifen treatment. Acute ablation of Smarcb1 resulted in a higher metastatic burden (n = 7 per group). Data are mean ± s.d. of biological replicates. k, Kaplan–Meier survival analysis of NCr Nude mice orthotopically transplanted with R26Cag-FlpoERT2/Cag-LSL-Luc- KPΔ/Δ-pLSM5-shSmarcb1-1 or R26Cag-FlpoERT2/Cag-LSL-Luc-KPΔ/Δ-pLSM5-shSmarcb1-855 tumours (n = 7 per group). Tamoxifen treatment was started 5 days after surgery. Acute ablation of Smarcb1 resulted in more aggressive tumours and a significantly shorter overall survival. l, Representative ex-vivo bioluminescence images in NCr Nude mice orthotopically transplanted with R26Cag-FlpoERT2/Cag-LSL-Luc-KPΔ/Δ-pLSM5-shSmarcb1-855 tumours and assigned to vehicle or tamoxifen treatment. Arrows indicate metastatic liver disease. m, Schematic showing the lentiviral construct for Cre-mediated, tissue-specific, time-restricted restoration of a gene of interest in vivo (pLSM2). High-titre-purified lentiviral particles (2–5 × 108 IU) are introduced surgically into the pancreas of KrasG12DFSF/+-Tp53Frt/Frt-R26CreERT2/+ mice to generate the R26CreERT2/+-KPFrt/Frt-pLSM2-shSmarcb1 model. 7 days after surgery, mice were treated with caerulein. The tissue specificity is provided by the human Krt19 promoter, which drives the expression of the Flpo recombinase, resulting in the activation of the latent mutant KrasG12DFSF/+ and the inactivation of the conditional Tp53Frt allele along with the constitutive expression of an shRNA under the U6 promoter. The time-restricted restoration of the gene of interest is mediated by the R26CreERT2 ubiquitous CreERT2 strain upon tamoxifen treatment and removal of the cassette containing the shRNA flanked by LoxP sites. n, Left, T2-weighted MRI scans of tumour-bearing mice 19 weeks after orthotopic injection with the pLSM2 lentiviral system carrying two shRNAs specific for murine Smarcb1. The tumours extensively invade the abdominal cavity. Right, immuno-phenotype of a tumour induced with pLSM2-shSmarcb1. Poorly cohesive, undifferentiated tumour cells express the pancreatic-specific markers Pdx1 and Sox9, suggestive of a pancreatic epithelial cell of origin. o, Kaplan–Meier analysis of tumour incidence in R26CreERT2/+-KPFrt/Frt mice challenged with orthotopic injections of the pLSM2-shSmarcb1 and pLSM2-shCtrl constructs. Knockdown of Smarcb1 in the mosaic model system results in higher penetrance and shorter latency (R26CreERT2/+-KPFrt/Frt-pLSM2-shSmarcb1, n = 53; R26CreERT2/+-KPFrt/Frt-pLSM2-shCtrl, n = 39). p, Quantification of the metastatic burden in R26CreERT2/+-KPFrt/Frt-pLSM2-shSmarcb1 mice versus R26CreERT2/+-KPFrt/Frt-pLSM2-shCtrl mice, assessed by counting the combined number of liver, lung and peritoneal metastases (R26CreERT2/+-KPFrt/Frt-pLSM2-shSmarcb1, n = 23; R26CreERT2/+-KPFrt/Frt-pLSM2-shCtrl, n = 5). Data are mean ± s.d. of biological replicates. q, PCR from recombined and un-recombined genomic DNA isolated from vehicle- and tamoxifen-treated tumour-bearing mice. Genomic DNA was extracted 5 and 10 days after treatment in vivo. r, Kaplan–Meier survival analysis of R26CreERT2/+-KPFrt/Frt-pLSM2-shSmarcb1 tumours transplanted orthotopically in syngeneic C57BL/6 recipient mice (n = 5 per vehicle and d18 groups, n = 7 per d1 group). Early tamoxifen treatment and Smarcb1 restoration resulted in a significant and durable improvement in survival. s, Metastatic burden was estimated by counting the number of liver, peritoneal and lung metastasis in the experimental cohorts described in r (n = 5 per vehicle and Txd18 groups, n = 7 per Txd1 group). The early restoration of Smarcb1 greatly reduced the number of metastatic foci, suggesting that its deficiency is a requirement in tumour dissemination. Data are mean ± s.d. of biological replicates. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, by Mantel–Cox log-rank test (c, k, o, r), two-tailed chi-squared test (h) or unpaired two-tailed t-test (i, j, p, s). Scale bars: 100 μm (b, d, f, n). For gel source data, see Supplementary Figures.