

Comparative analysis estimates the relative frequencies of co-divergence and cross-species transmission within viral families
- PMID: 28178344
- PMCID: PMC5319820
- DOI: 10.1371/journal.ppat.1006215
Comparative analysis estimates the relative frequencies of co-divergence and cross-species transmission within viral families
Abstract
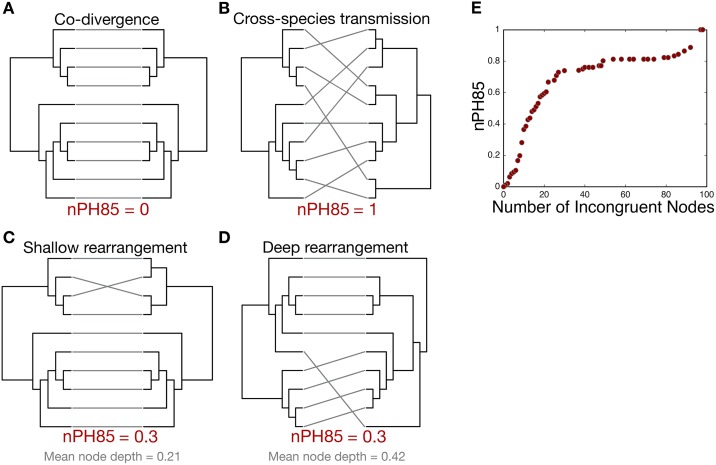
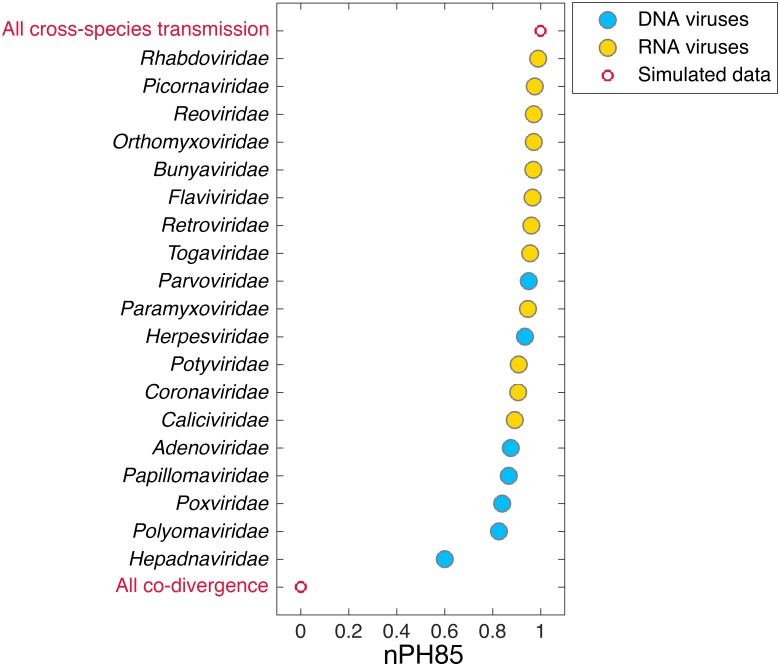
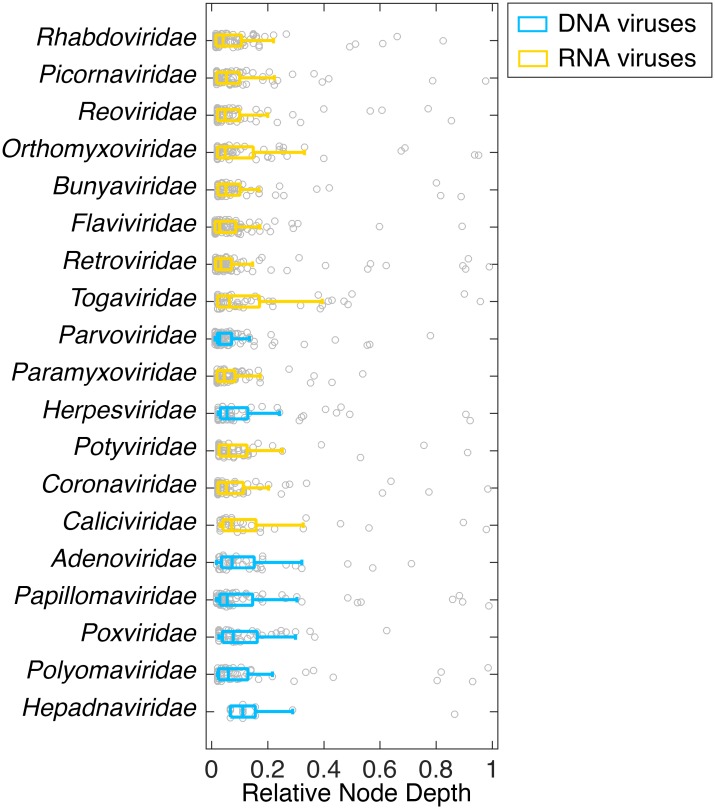
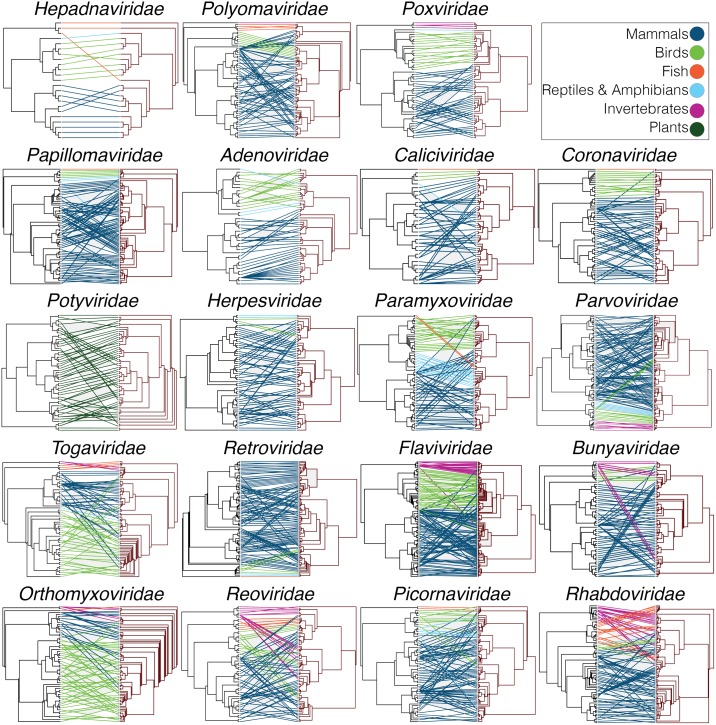
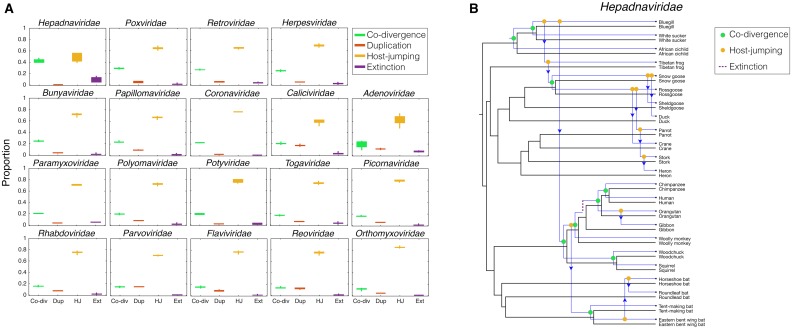
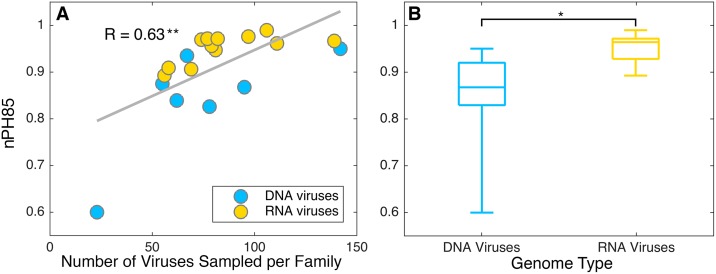
The cross-species transmission of viruses from one host species to another is responsible for the majority of emerging infections. However, it is unclear whether some virus families have a greater propensity to jump host species than others. If related viruses have an evolutionary history of co-divergence with their hosts there should be evidence of topological similarities between the virus and host phylogenetic trees, whereas host jumping generates incongruent tree topologies. By analyzing co-phylogenetic processes in 19 virus families and their eukaryotic hosts we provide a quantitative and comparative estimate of the relative frequency of virus-host co-divergence versus cross-species transmission among virus families. Notably, our analysis reveals that cross-species transmission is a near universal feature of the viruses analyzed here, with virus-host co-divergence occurring less frequently and always on a subset of viruses. Despite the overall high topological incongruence among virus and host phylogenies, the Hepadnaviridae, Polyomaviridae, Poxviridae, Papillomaviridae and Adenoviridae, all of which possess double-stranded DNA genomes, exhibited more frequent co-divergence than the other virus families studied here. At the other extreme, the virus and host trees for all the RNA viruses studied here, particularly the Rhabdoviridae and the Picornaviridae, displayed high levels of topological incongruence, indicative of frequent host switching. Overall, we show that cross-species transmission plays a major role in virus evolution, with all the virus families studied here having the potential to jump host species, and that increased sampling will likely reveal more instances of host jumping.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Holmes EC. Evolution and emergence of RNA viruses: Oxford University Press; 2009.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
