

Half a Century Tales of Familial Hypercholesterolemia (FH) in Japan
- PMID: 28179607
- PMCID: PMC5383536
- DOI: 10.5551/jat.RV16008
Half a Century Tales of Familial Hypercholesterolemia (FH) in Japan
Abstract
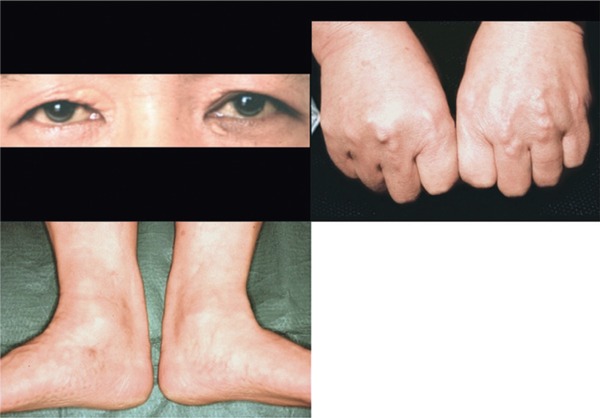
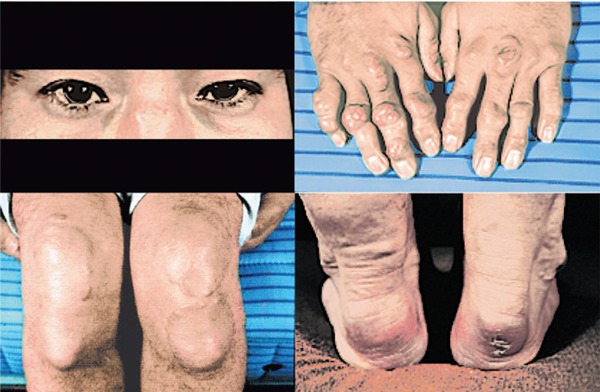
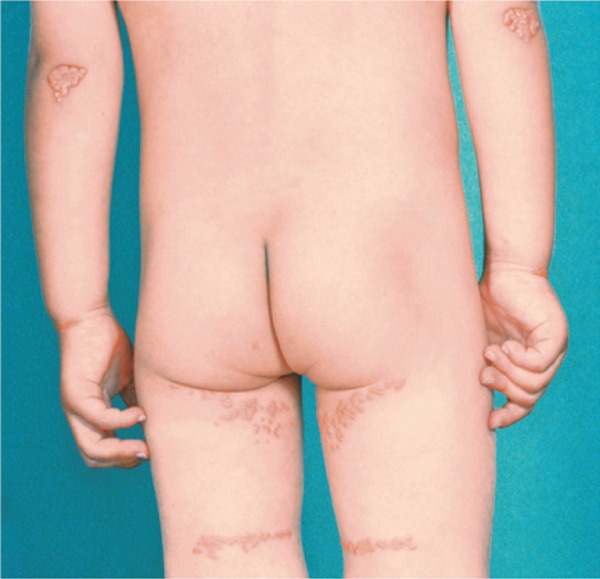
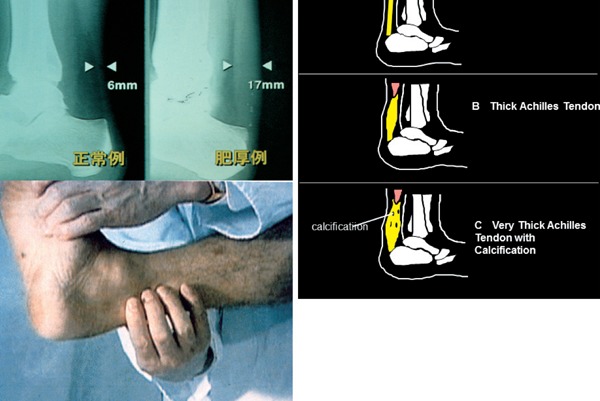
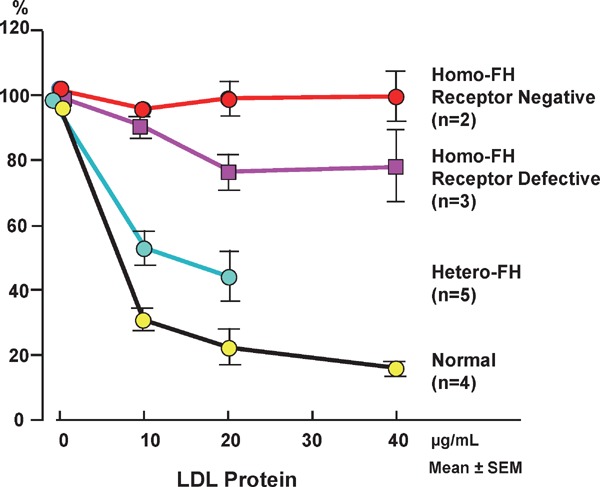
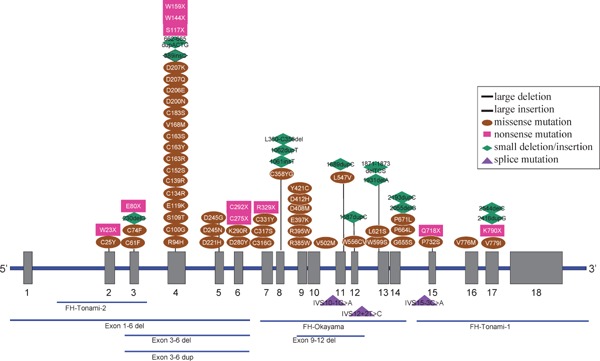
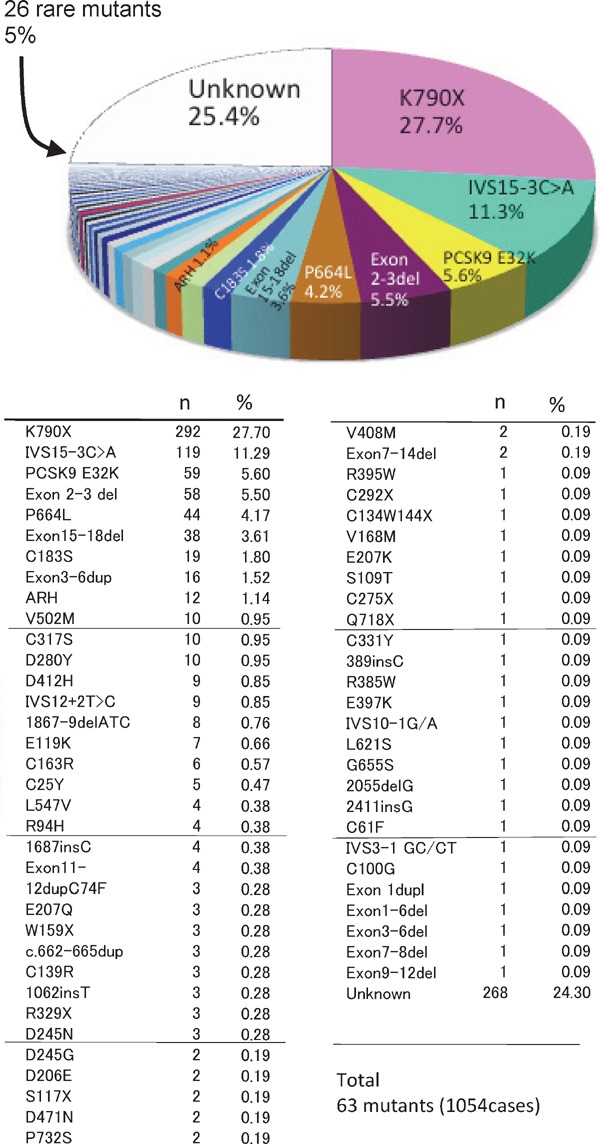
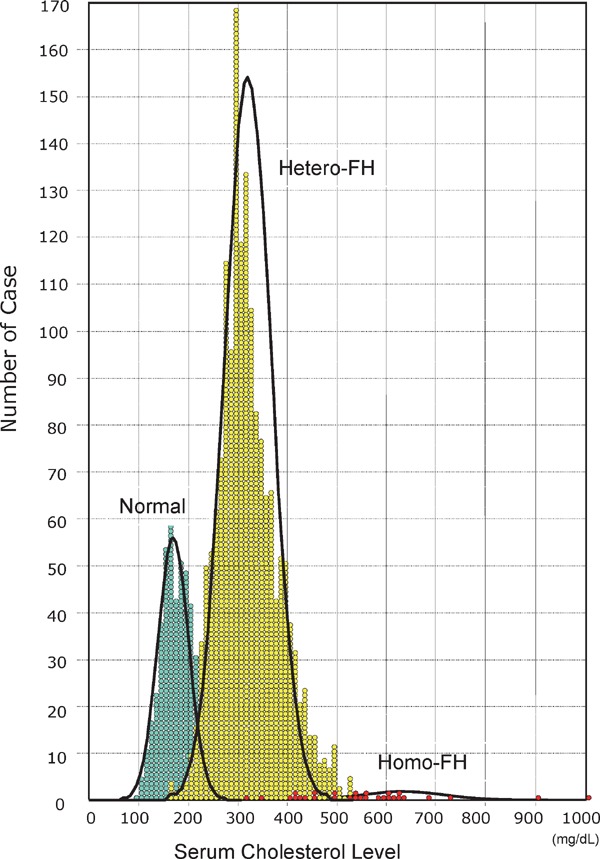
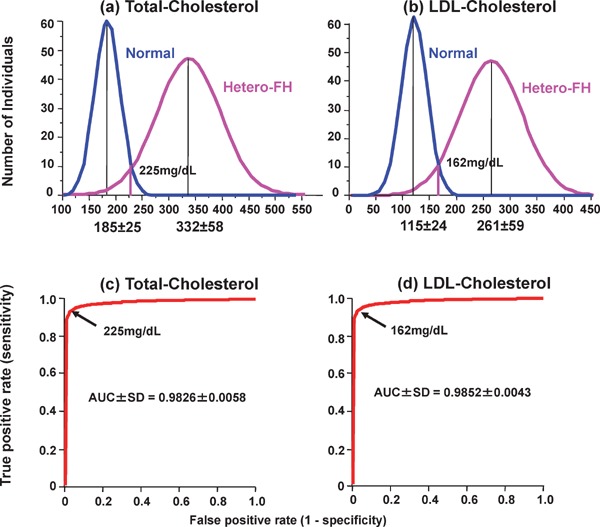
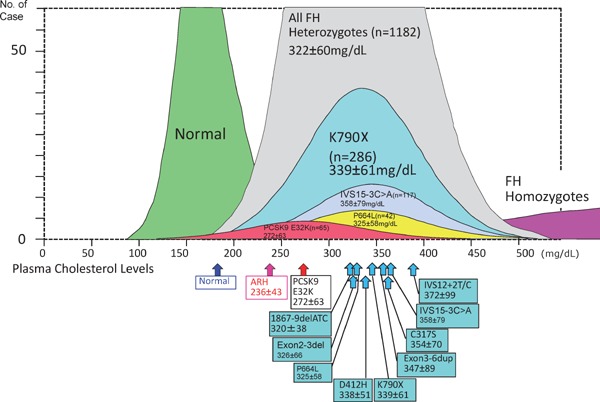
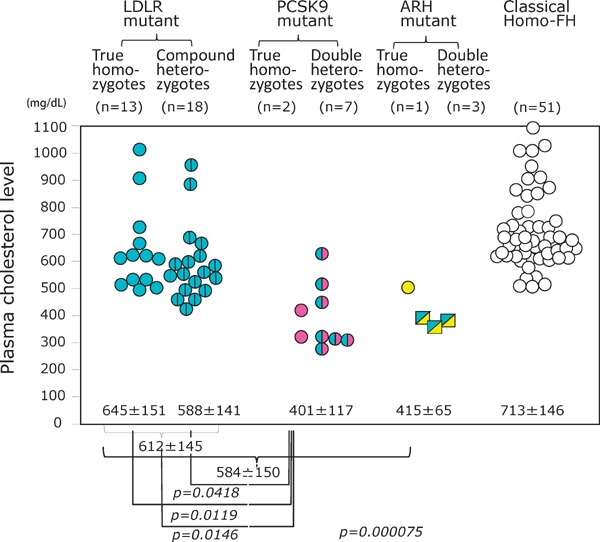
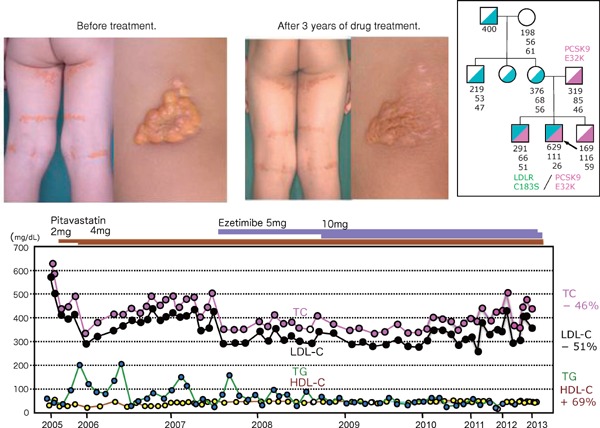
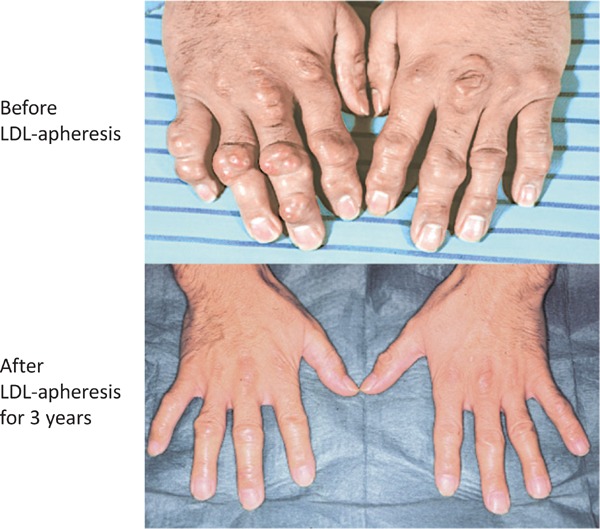
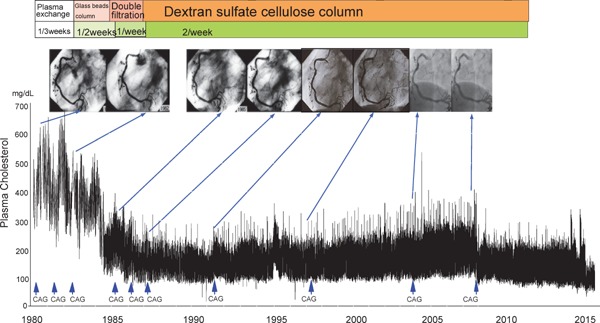
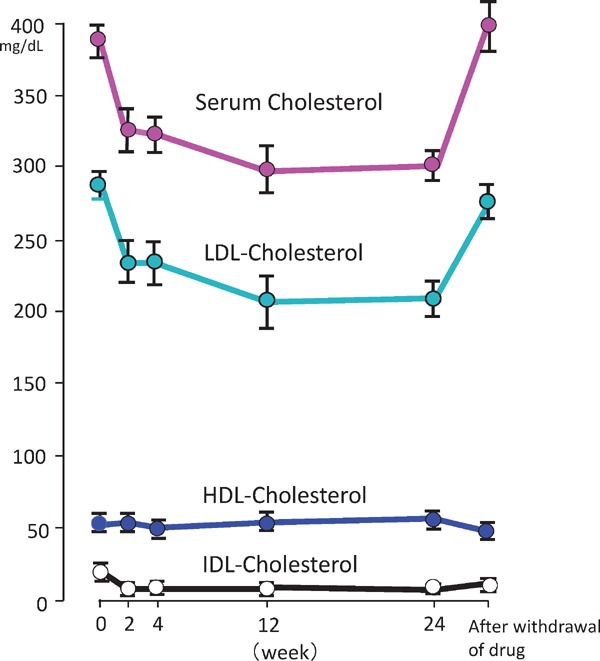
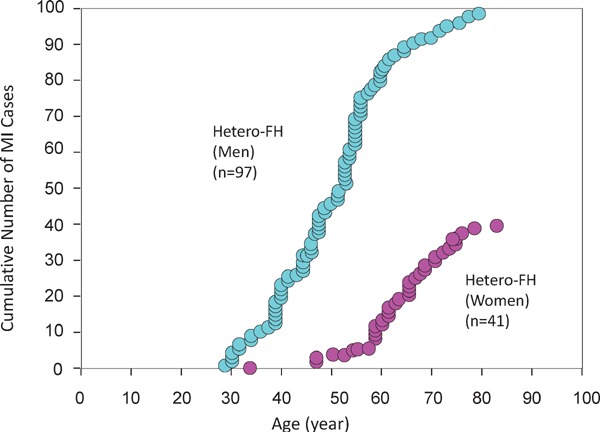
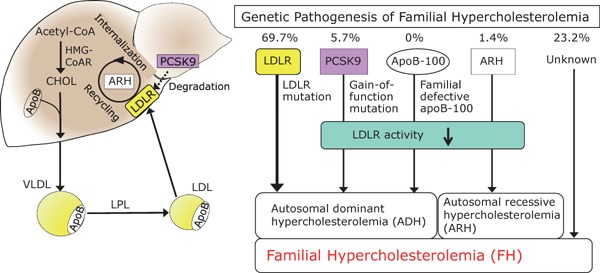
Familial hypercholesterolemia (FH) is a disease characterized by a triad: elevated low-density lipoprotein (LDL) cholesterol, tendon xanthomas, and premature coronary heart disease. Thus, it can be considered as a model disease for hypercholesterolemia and atherosclerotic cardiovascular disease (ASCVD). For the diagnosis of hetero-FH, the detection of Achilles tendon xanthomas by palpation or on X-ray is an indispensable diagnostic skill in clinical lipidology. To prevent the under-diagnosis and under-treatment of FH, the diagnostic criteria should be more convenient and user-friendly. For a patient with cutaneous or tendon xanthomas, the probability of FH is very high; however, an absence of xanthoma does not rule out FH.Brown and Goldstein elucidated the pathogenesis of FH by their work on LDL-receptor (LDL-R), for which they were awarded the Nobel Prize in 1985. In the 1950s, FH patients were divided into heterozygous (hetero-) and homozygous (homo-) FH, and diagnosing homo- and hetero-FH based on the phenotypic features of ASCVD or xanthomas frequently became difficult without the DNA analysis of FH genes. It is estimated that heterozygous mutations in the LDL-R or the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene will be found at a combined frequency of 0.005, which corresponds to 1/199 people in the general population in Japan.Statins and anti-PCSK9 monoclonal antibodies are highly specific and efficient drugs for treating hetero- or homo-FH patients. Most clinical studies have reported an amelioration of ASCVD using long-term statin therapy. Clinical results using anti-PCSK9 monoclonal antibodies will emerge in a few years. In homo-FH patients, mipomersen and lomitapide are expected to yield good results. It is important to sequentially unravel the unrecognized pathogenetic mechanisms of FH to reduce its under-recognition and develop new management strategies for it.
Conflict of interest statement
H.M. has no conflict of interest concerning this publication.
Figures


References
-
- Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In the Metabolic and molecular bases of inherited disease. ed. by Scriver C, Beaudet A, Sly W, Valle D, Childs B, Kinzler K, Vogelstein B. McGraw-Hill Professional; 8th ed. 2000
-
- Fagge CH. Xanthomatous disease of the skin. I. General xanthelasma or vitiligoides. Trans Pathol Soc Lond 24: 242-250, 1873
-
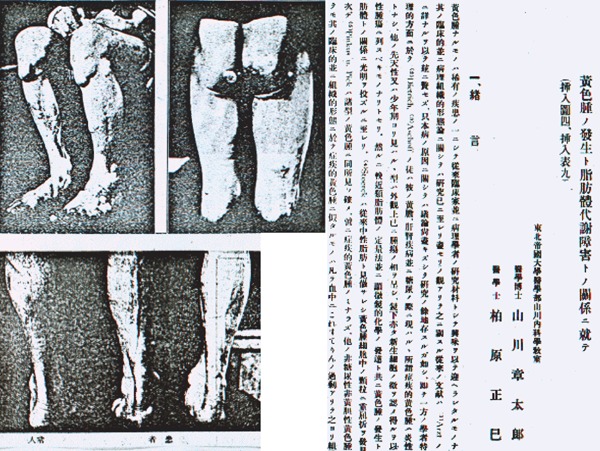
- Yamakawa S, Kashiwabara M. Relashionship between pathogenesis of xanthoma and disturbance of lipid metabolism. (in Japanese). Tohoku Igakuzasshi 6: 322-336, 1922
-
- Mabuchi H, Tatami R, Haba T, Ueda K, Ueda R, Kametani T, Itoh S, Koizumi J, Oota M, Miyamoto S, Takeda R, Takeshita H. Homozygous familial hypercholesterolemia in Japan. Am J Med. 65: 290-297, 1978 - PubMed
-
- Fredrickson DS, Levy RI, Lees RS. Fat transport in lipoproteins - an integrated approach to mechanisms and disorders. N Engl J Med. 276: 34-42, 94,-103, 148,-156, 215,-225, 273-281, 1967 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous