

Ciliopathies: Genetics in Pediatric Medicine
- PMID: 28180024
- PMCID: PMC5289266
- DOI: 10.1055/s-0036-1593841
Ciliopathies: Genetics in Pediatric Medicine
Abstract
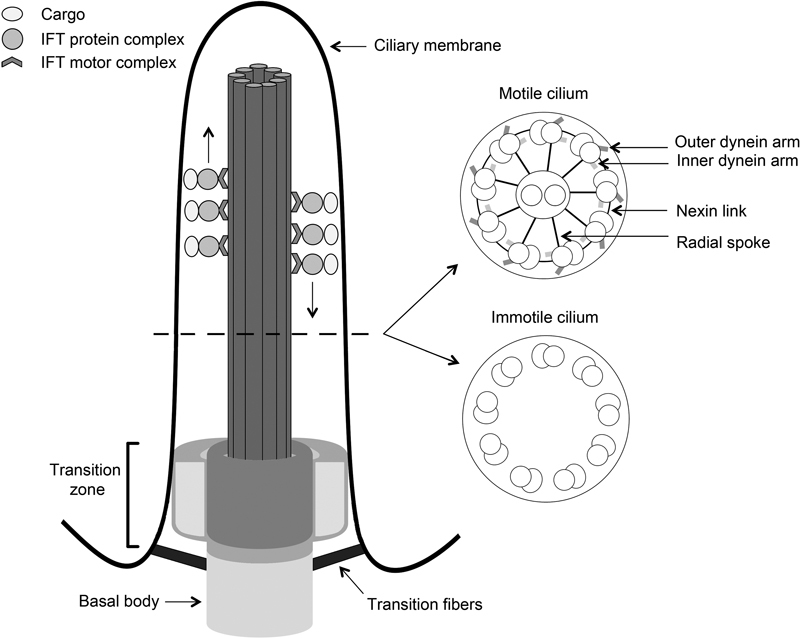
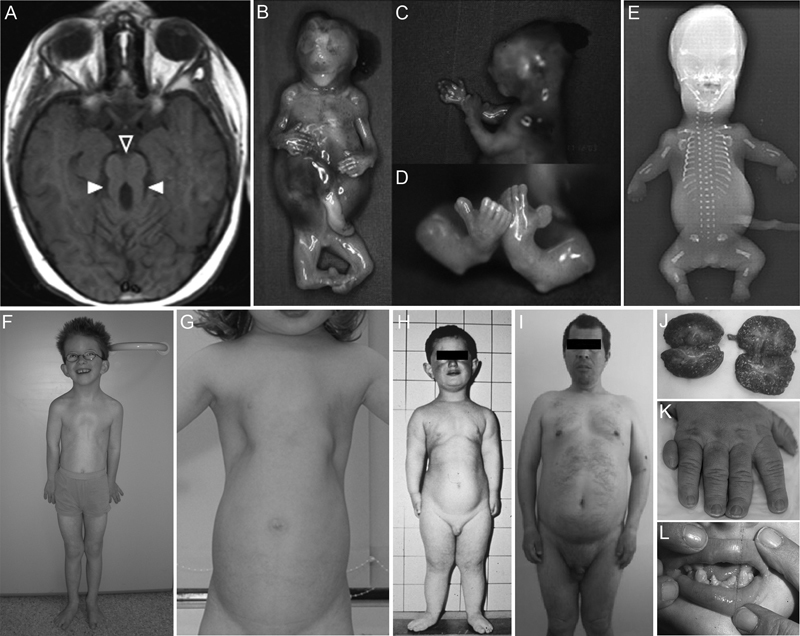
Ciliary disorders, which are also referred to as ciliopathies, are a group of hereditary disorders that result from dysfunctional cilia. The latter are cellular organelles that stick up from the apical plasma membrane. Cilia have important roles in signal transduction and facilitate communications between cells and their surroundings. Ciliary disruption can result in a wide variety of clinically and genetically heterogeneous disorders with overlapping phenotypes. Because cilia occur widespread in our bodies many organs and sensory systems can be affected when they are dysfunctional. Ciliary disorders may be isolated or syndromic, and common features are cystic liver and/or kidney disease, blindness, neural tube defects, brain anomalies and intellectual disability, skeletal abnormalities ranging from polydactyly to abnormally short ribs and limbs, ectodermal defects, obesity, situs inversus, infertility, and recurrent respiratory tract infections. In this review, we summarize the features, frequency, morbidity, and mortality of each of the different ciliopathies that occur in pediatrics. The importance of genetics and the occurrence of genotype-phenotype correlations are indicated, and advances in gene identification are discussed. The use of next-generation sequencing by which a gene panel or all genes can be screened in a single experiment is highlighted as this technology significantly lowered costs and time of the mutation detection process in the past. We discuss the challenges of this new technology and briefly touch upon the use of whole-exome sequencing as a diagnostic test for ciliary disorders. Finally, a perspective on the future of genetics in the context of ciliary disorders is provided.
Keywords: cilia; ciliopathy; diagnostics; genotype-phenotype; next-generation sequencing.
Figures
References
-
- Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8(11):880–893. - PubMed
-
- Satir P, Christensen S T. Overview of structure and function of mammalian cilia. Annu Rev Physiol. 2007;69:377–400. - PubMed
-
- Salathe M. Regulation of mammalian ciliary beating. Annu Rev Physiol. 2007;69:401–422. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
