

Small genomic insertions form enhancers that misregulate oncogenes
- PMID: 28181482
- PMCID: PMC5309821
- DOI: 10.1038/ncomms14385
Small genomic insertions form enhancers that misregulate oncogenes
Erratum in
-
Corrigendum: Small genomic insertions form enhancers that misregulate oncogenes.Nat Commun. 2017 Jun 1;8:15797. doi: 10.1038/ncomms15797. Nat Commun. 2017. PMID: 28569765 Free PMC article.
Abstract
The non-coding regions of tumour cell genomes harbour a considerable fraction of total DNA sequence variation, but the functional contribution of these variants to tumorigenesis is ill-defined. Among these non-coding variants, somatic insertions are among the least well characterized due to challenges with interpreting short-read DNA sequences. Here, using a combination of Chip-seq to enrich enhancer DNA and a computational approach with multiple DNA alignment procedures, we identify enhancer-associated small insertion variants. Among the 102 tumour cell genomes we analyse, small insertions are frequently observed in enhancer DNA sequences near known oncogenes. Further study of one insertion, somatically acquired in primary leukaemia tumour genomes, reveals that it nucleates formation of an active enhancer that drives expression of the LMO2 oncogene. The approach described here to identify enhancer-associated small insertion variants provides a foundation for further study of these abnormalities across human cancers.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
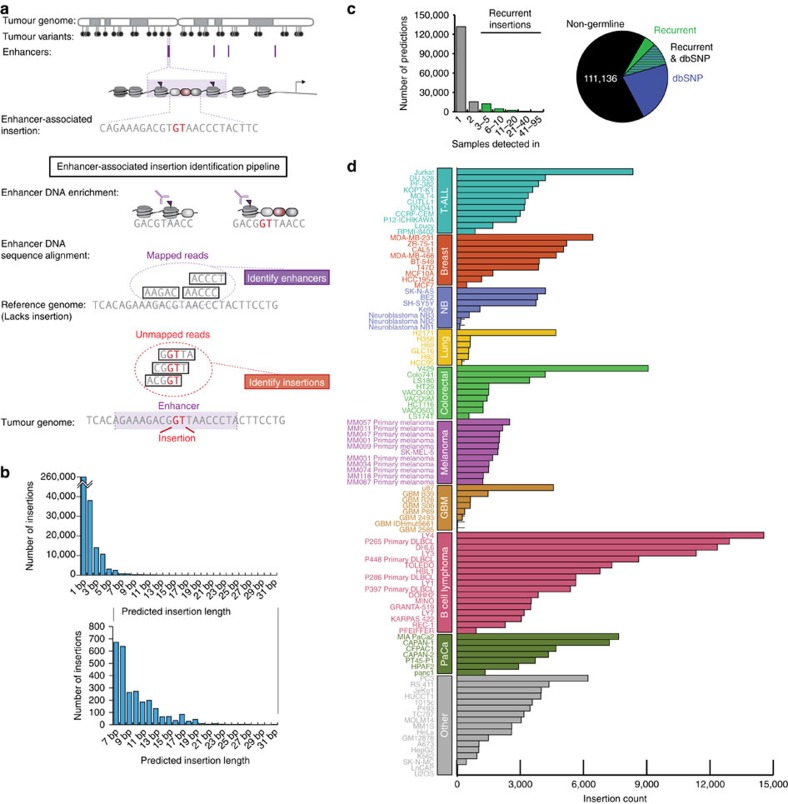
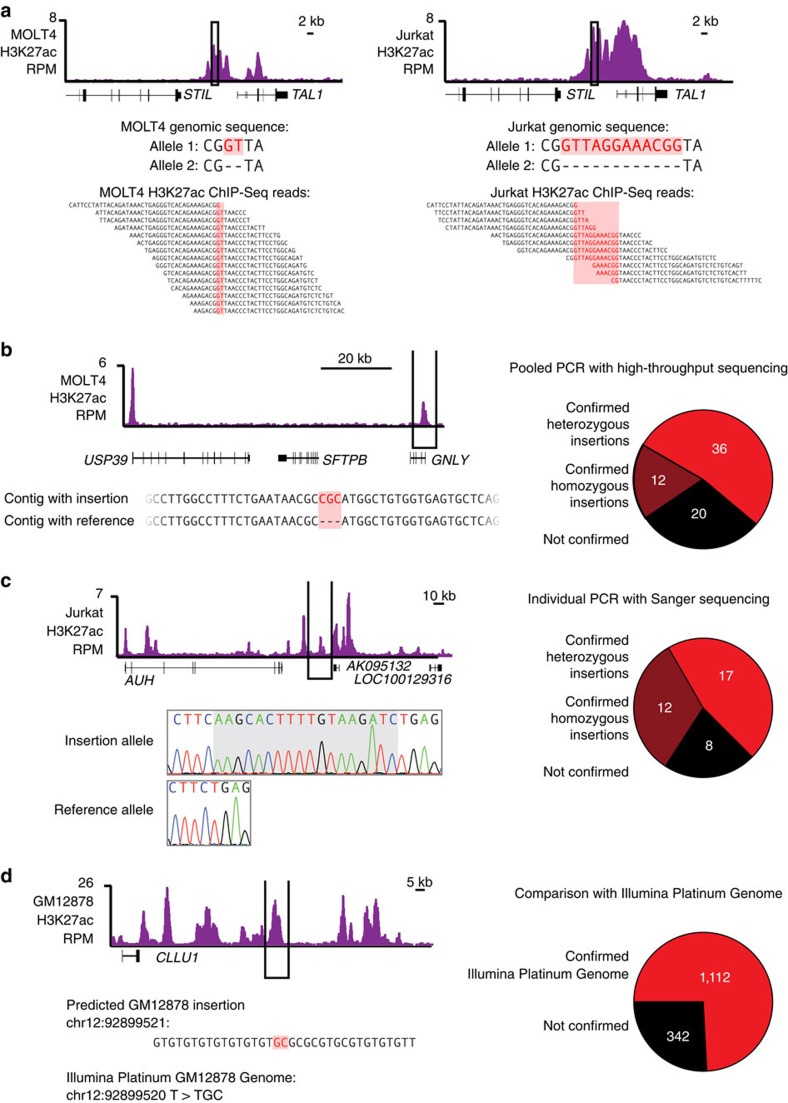
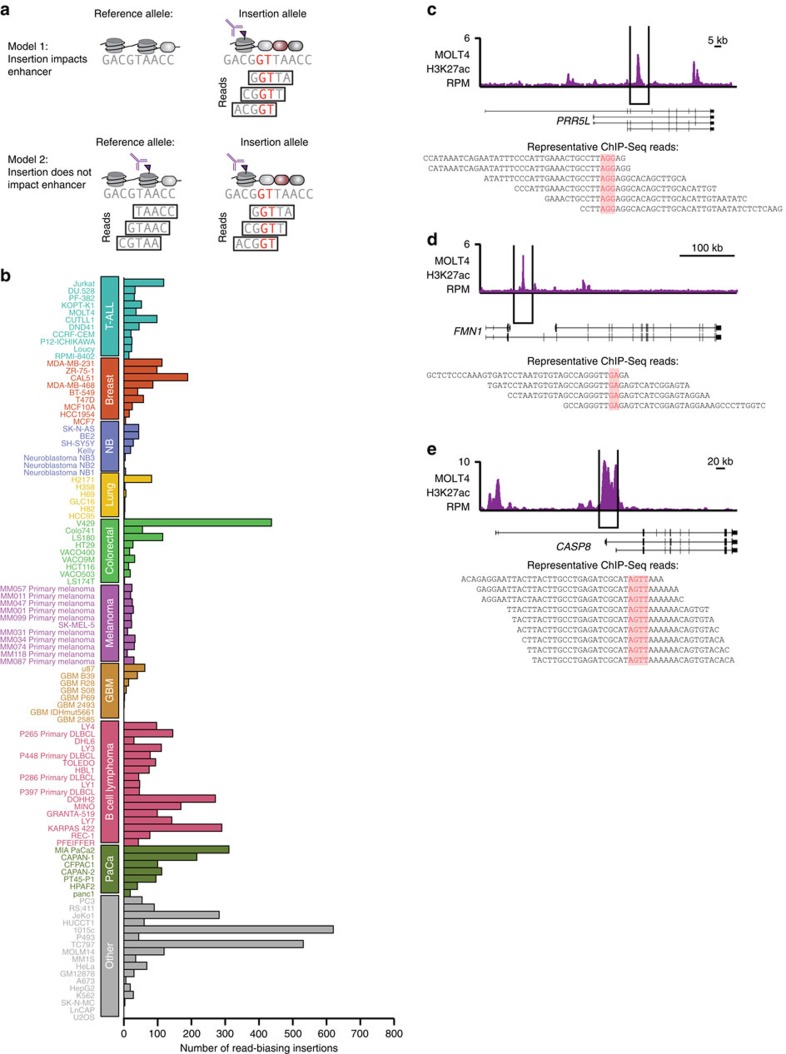
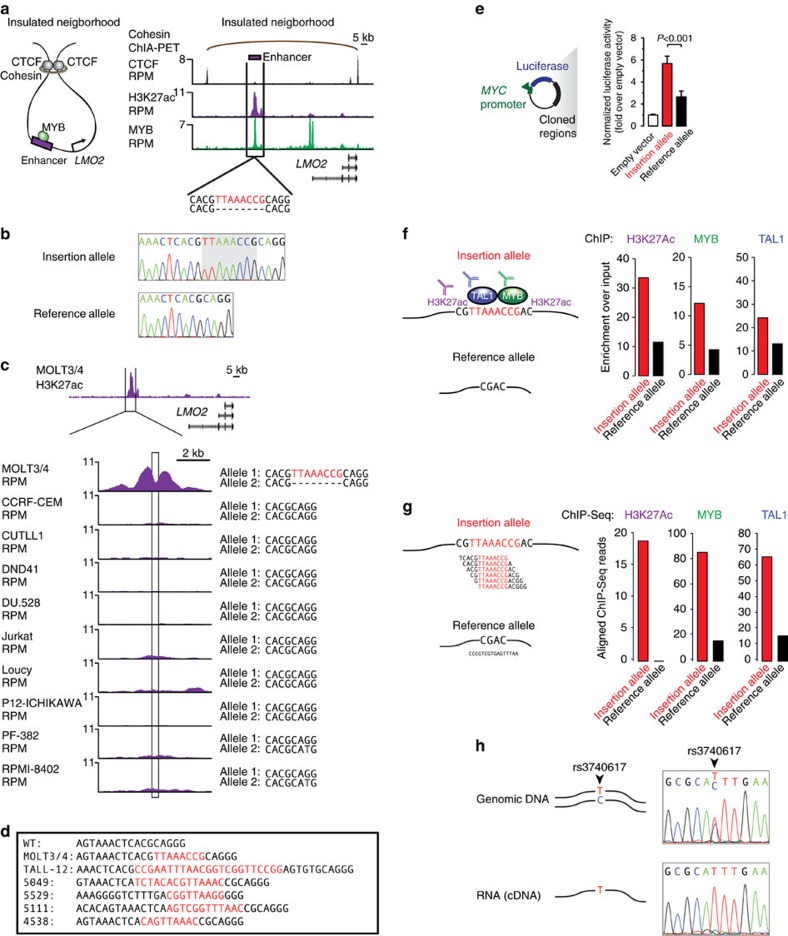
References
-
- Garraway L. A. & Lander E. S. Lessons from the cancer genome. Cell 153, 17–37 (2013). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
