

The far-reaching scope of neuroinflammation after traumatic brain injury
- PMID: 28186177
- PMCID: PMC5675525
- DOI: 10.1038/nrneurol.2017.13
The far-reaching scope of neuroinflammation after traumatic brain injury
Erratum in
-
The far-reaching scope of neuroinflammation after traumatic brain injury.Nat Rev Neurol. 2017 Sep;13(9):572. doi: 10.1038/nrneurol.2017.116. Epub 2017 Aug 4. Nat Rev Neurol. 2017. PMID: 28776601
Abstract
The 'silent epidemic' of traumatic brain injury (TBI) has been placed in the spotlight as a result of clinical investigations and popular press coverage of athletes and veterans with single or repetitive head injuries. Neuroinflammation can cause acute secondary injury after TBI, and has been linked to chronic neurodegenerative diseases; however, anti-inflammatory agents have failed to improve TBI outcomes in clinical trials. In this Review, we therefore propose a new framework of targeted immunomodulation after TBI for future exploration. Our framework incorporates factors such as the time from injury, mechanism of injury, and secondary insults in considering potential treatment options. Structuring our discussion around the dynamics of the immune response to TBI - from initial triggers to chronic neuroinflammation - we consider the ability of soluble and cellular inflammatory mediators to promote repair and regeneration versus secondary injury and neurodegeneration. We summarize both animal model and human studies, with clinical data explicitly defined throughout this Review. Recent advances in neuroimmunology and TBI-responsive neuroinflammation are incorporated, including concepts of inflammasomes, mechanisms of microglial polarization, and glymphatic clearance. Moreover, we highlight findings that could offer novel therapeutic targets for translational and clinical research, assimilate evidence from other brain injury models, and identify outstanding questions in the field.
Conflict of interest statement
The authors declare no competing interests
Figures
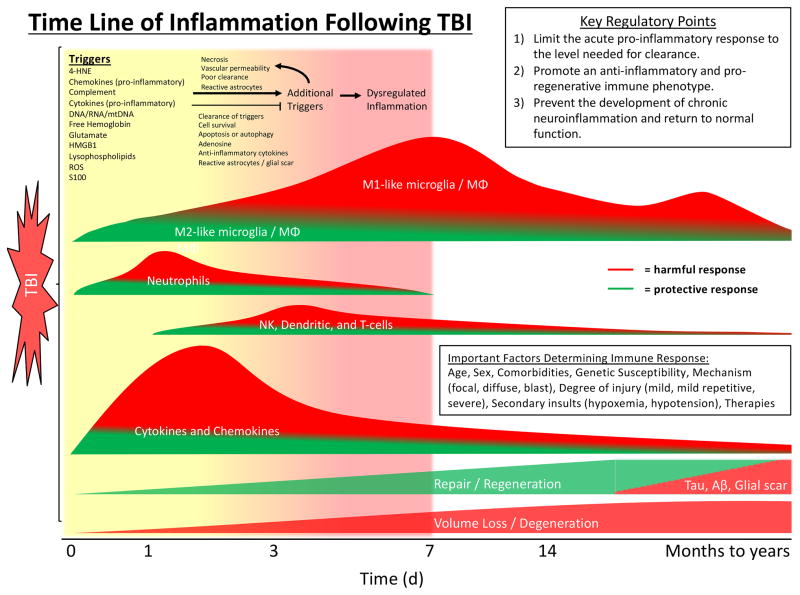
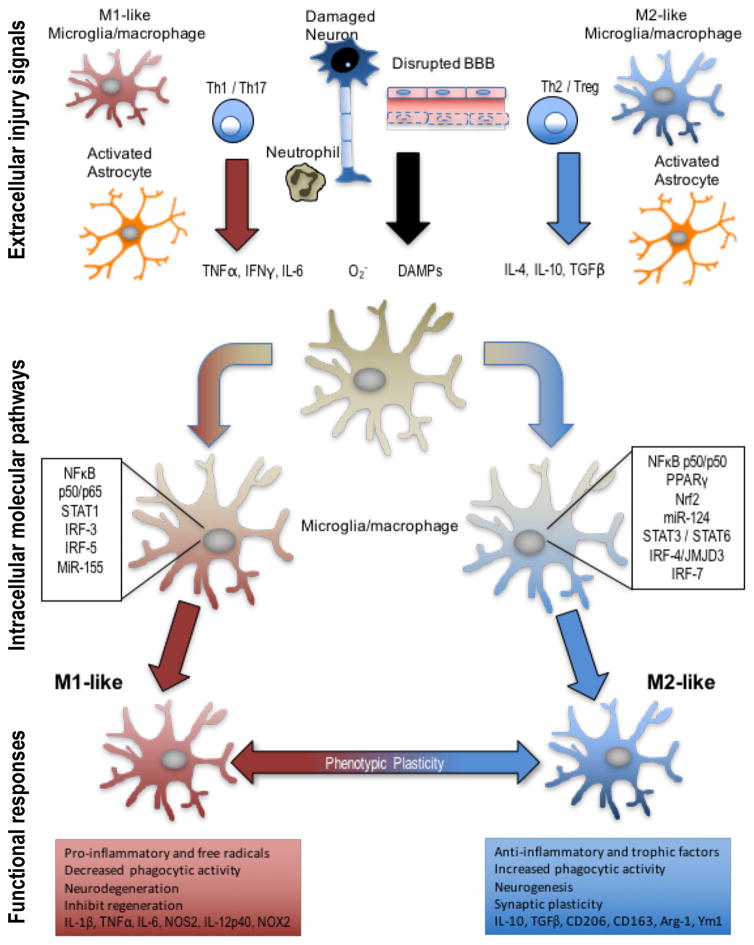
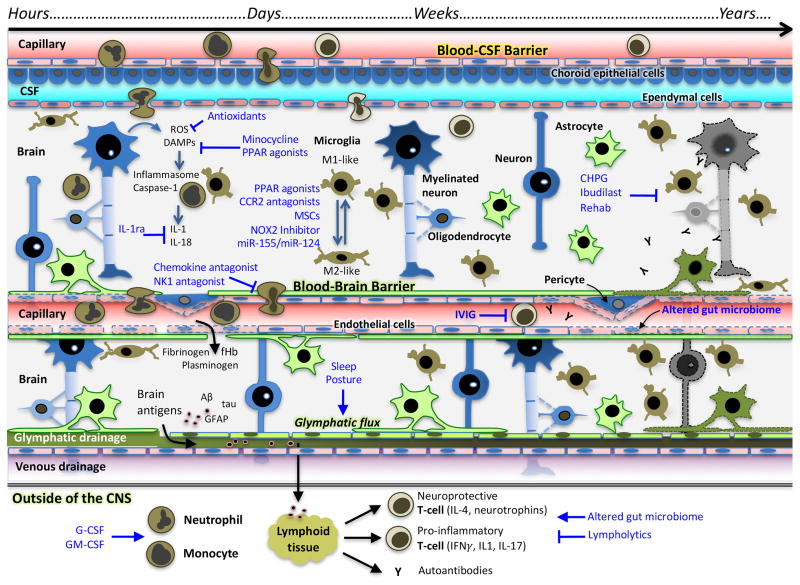
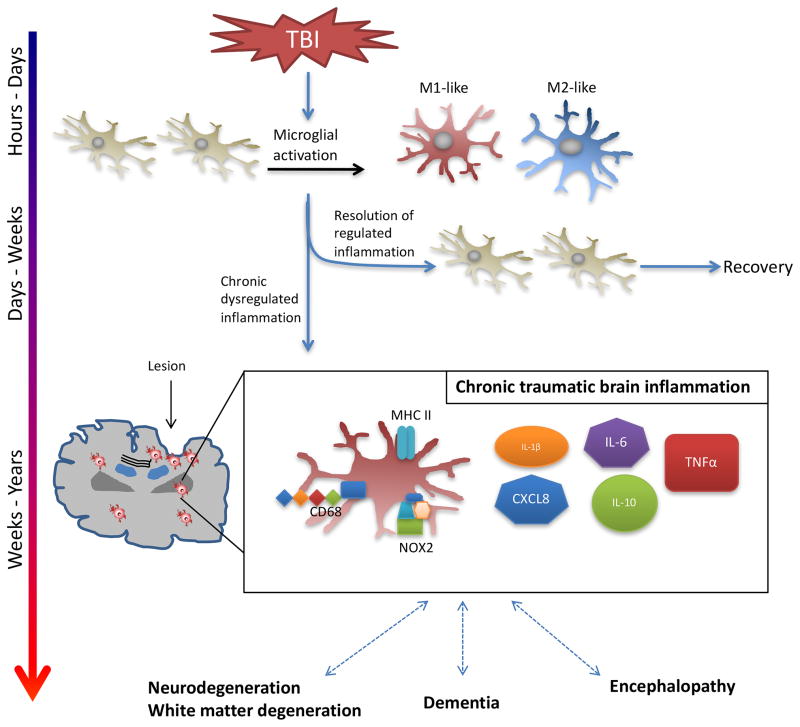
References
-
- Csuka E, et al. IL-10 levels in cerebrospinal fluid and serum of patients with severe traumatic brain injury: relationship to IL-6, TNF-alpha, TGF-beta1 and blood-brain barrier function. Journal of neuroimmunology. 1999;101:211–221. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
