

Sputum DNA sequencing in cystic fibrosis: non-invasive access to the lung microbiome and to pathogen details
- PMID: 28187782
- PMCID: PMC5303297
- DOI: 10.1186/s40168-017-0234-1
Sputum DNA sequencing in cystic fibrosis: non-invasive access to the lung microbiome and to pathogen details
Abstract
Background: Cystic fibrosis (CF) is a life-threatening genetic disorder, characterized by chronic microbial lung infections due to abnormally viscous mucus secretions within airways. The clinical management of CF typically involves regular respiratory-tract cultures in order to identify pathogens and to guide treatment. However, culture-based methods can miss atypical or slow-growing microbes. Furthermore, the isolated microbes are often not classified at the strain level due to limited taxonomic resolution.
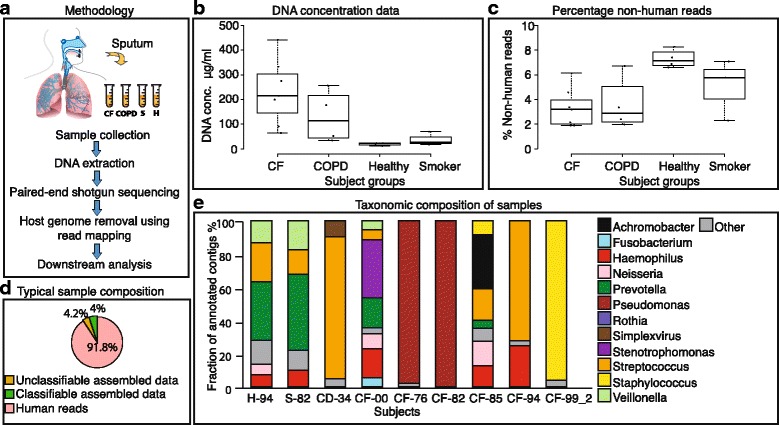
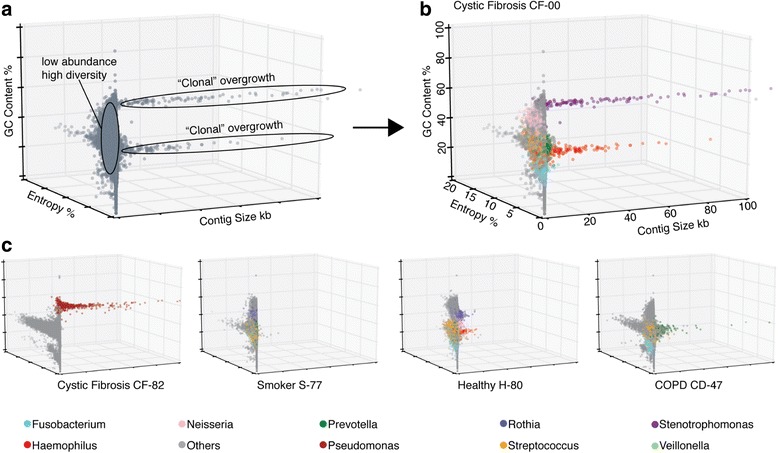
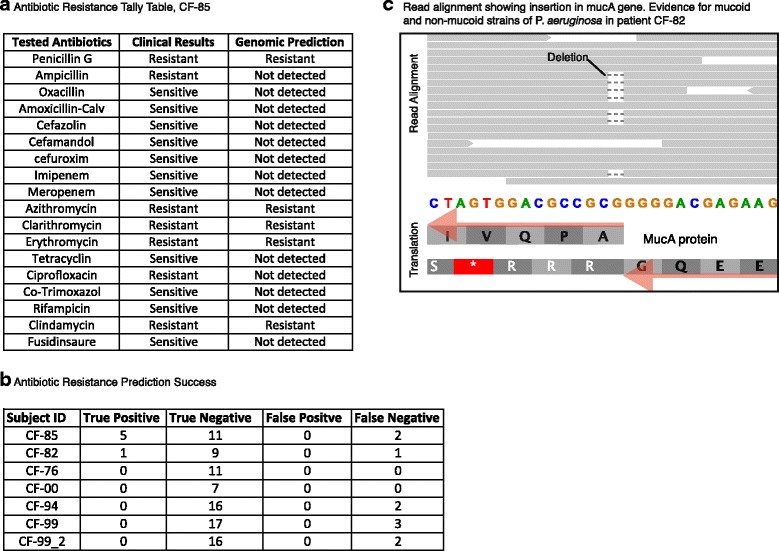
Results: Here, we show that untargeted metagenomic sequencing of sputum DNA can provide valuable information beyond the possibilities of culture-based diagnosis. We sequenced the sputum of six CF patients and eleven control samples (including healthy subjects and chronic obstructive pulmonary disease patients) without prior depletion of human DNA or cell size selection, thus obtaining the most unbiased and comprehensive characterization of CF respiratory tract microbes to date. We present detailed descriptions of the CF and healthy lung microbiome, reconstruct near complete pathogen genomes, and confirm that the CF lungs consistently exhibit reduced microbial diversity. Crucially, the obtained genomic sequences enabled a detailed identification of the exact pathogen strain types, when analyzed in conjunction with existing multi-locus sequence typing databases. We also detected putative pathogenicity islands and indicators of antibiotic resistance, in good agreement with independent clinical tests.
Conclusions: Unbiased sputum metagenomics provides an in-depth profile of the lung pathogen microbiome, which is complementary to and more detailed than standard culture-based reporting. Furthermore, functional and taxonomic features of the dominant pathogens, including antibiotics resistances, can be deduced-supporting accurate and non-invasive clinical diagnosis.
Keywords: COPD; Cystic fibrosis; Lung metagenome; Sputum; WGS metagenomic sequencing.
Figures


References
-
- Walters S, Mehta A. Epidemiology of cystic fibrosis. In: Hodson M, Geddes DM, Bush A, editors. Cystic fibrosis, 3rd edn. London: Edward Arnold Ltd; 2007. p. 21–45.
-
- Mahenthiralingam E. Emerging cystic fibrosis pathogens and the microbiome. Paediatr Respir Rev. 2014;15:13–5. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
