

Construction and Analysis of Two Genome-Scale Deletion Libraries for Bacillus subtilis
- PMID: 28189581
- PMCID: PMC5400513
- DOI: 10.1016/j.cels.2016.12.013
Construction and Analysis of Two Genome-Scale Deletion Libraries for Bacillus subtilis
Abstract
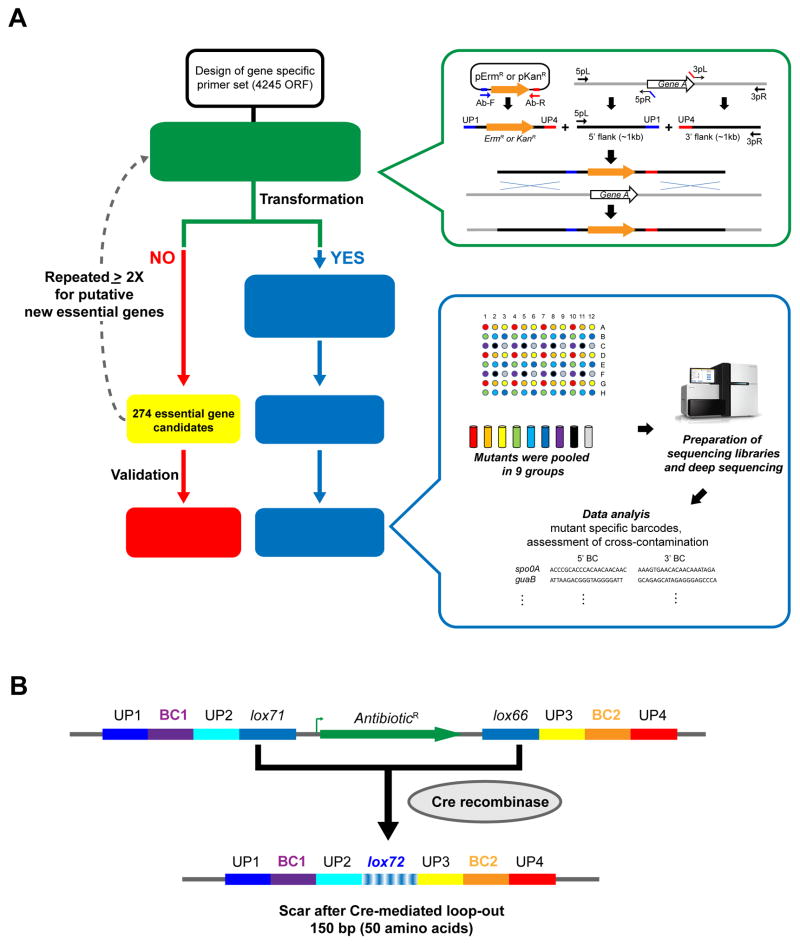
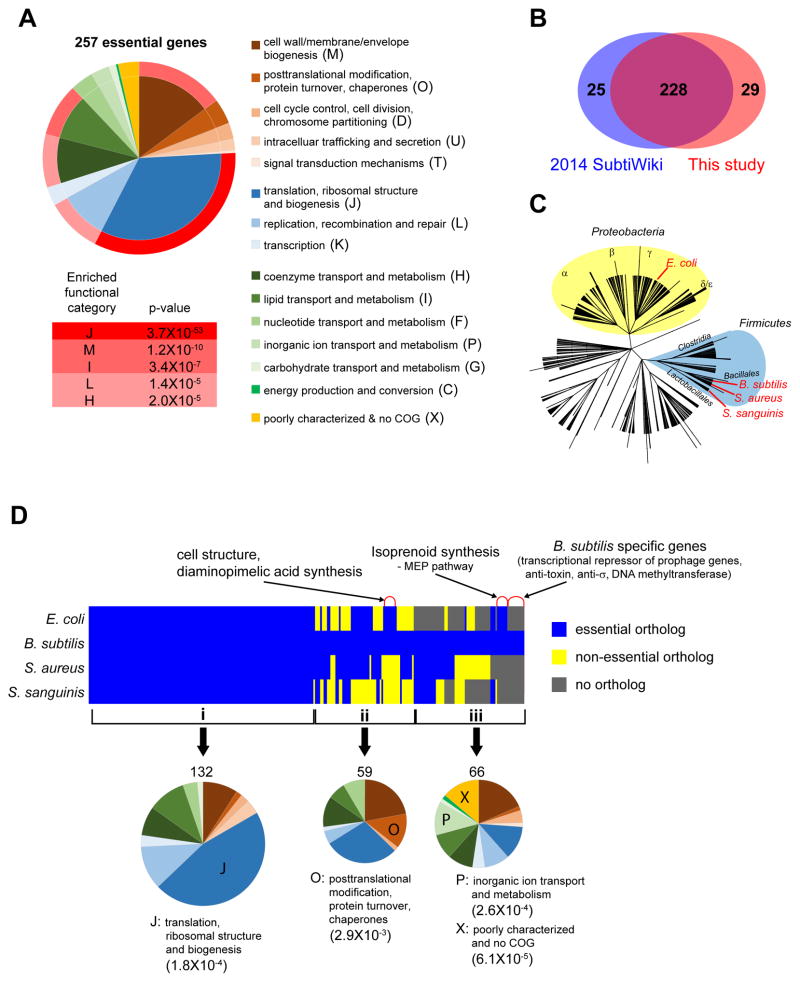
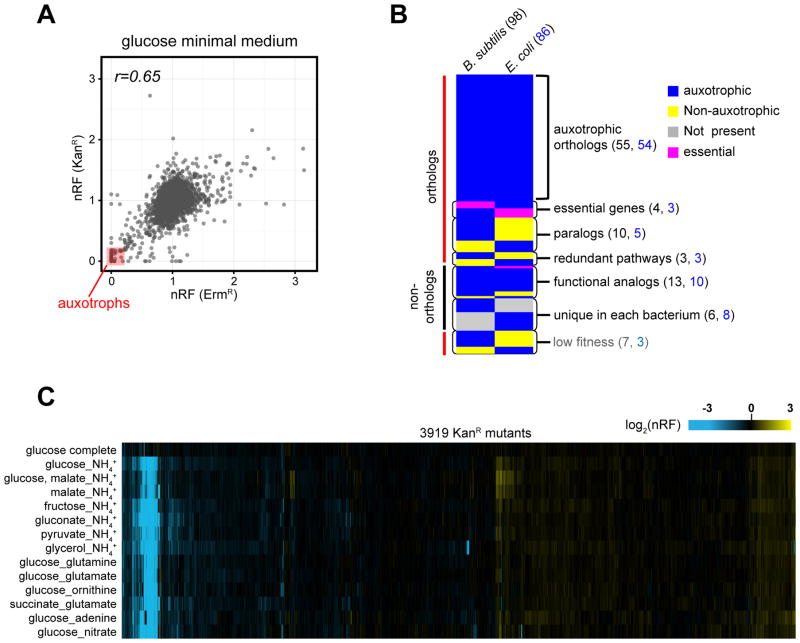
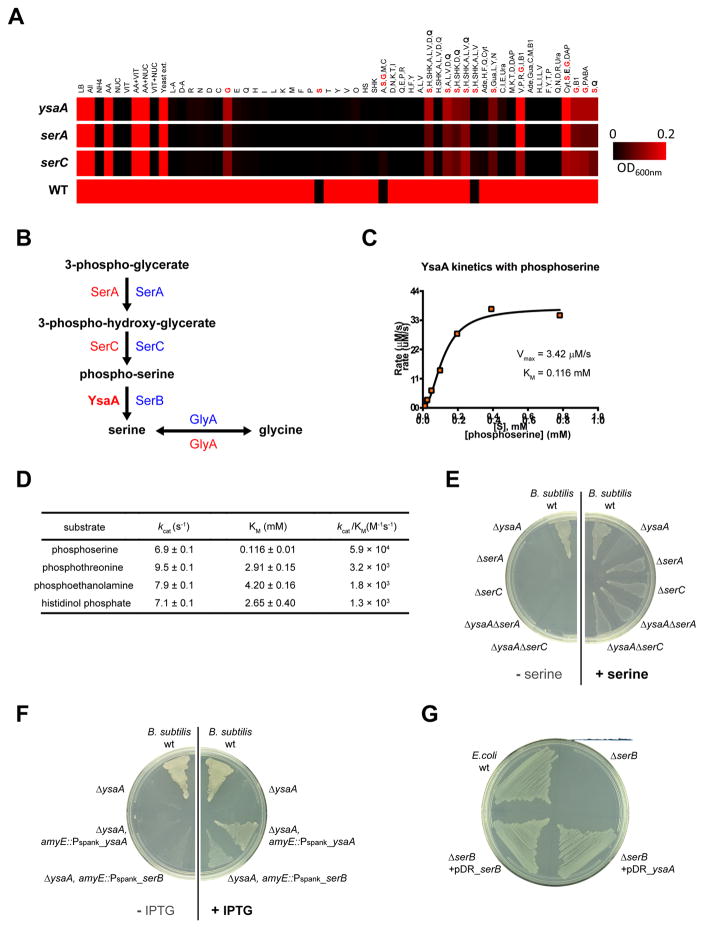
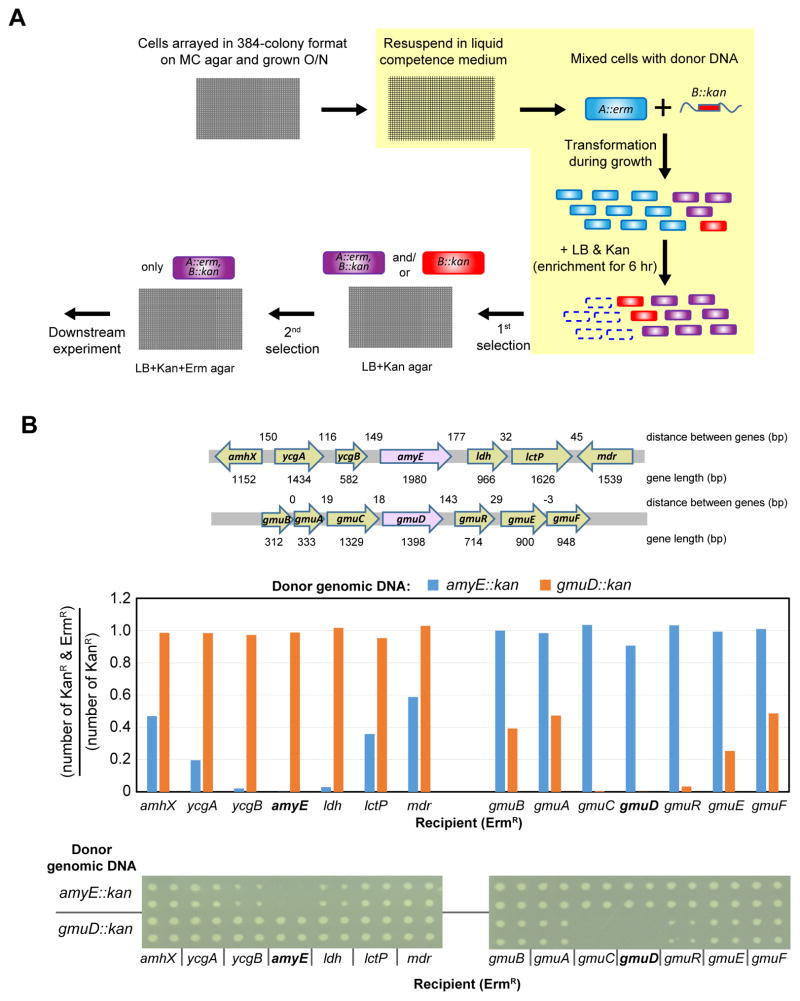
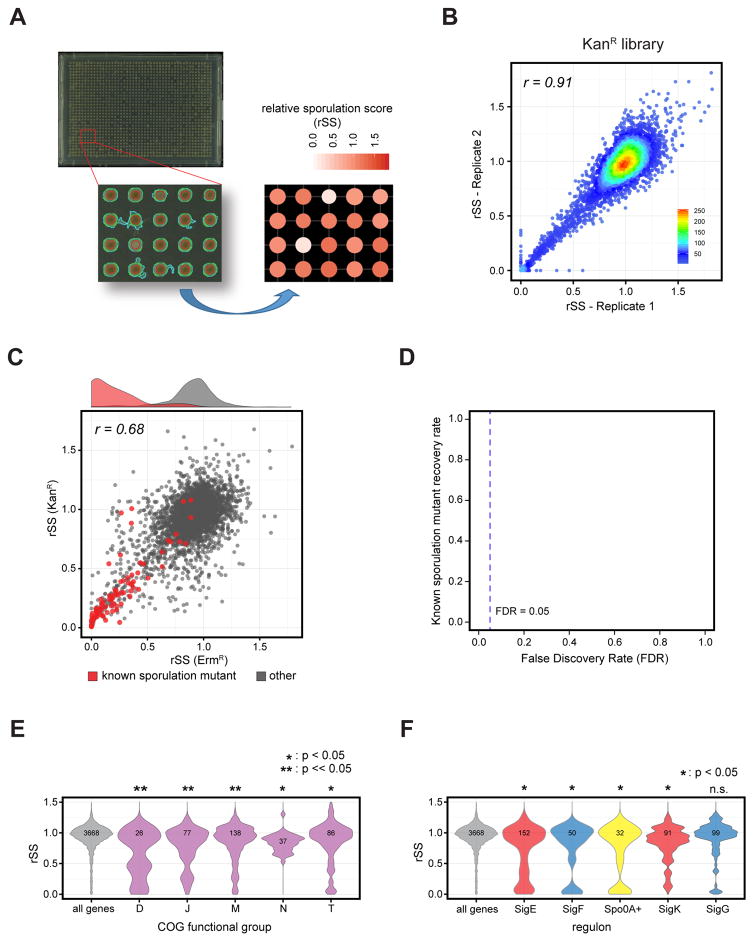
A systems-level understanding of Gram-positive bacteria is important from both an environmental and health perspective and is most easily obtained when high-quality, validated genomic resources are available. To this end, we constructed two ordered, barcoded, erythromycin-resistance- and kanamycin-resistance-marked single-gene deletion libraries of the Gram-positive model organism, Bacillus subtilis. The libraries comprise 3,968 and 3,970 genes, respectively, and overlap in all but four genes. Using these libraries, we update the set of essential genes known for this organism, provide a comprehensive compendium of B. subtilis auxotrophic genes, and identify genes required for utilizing specific carbon and nitrogen sources, as well as those required for growth at low temperature. We report the identification of enzymes catalyzing several missing steps in amino acid biosynthesis. Finally, we describe a suite of high-throughput phenotyping methodologies and apply them to provide a genome-wide analysis of competence and sporulation. Altogether, we provide versatile resources for studying gene function and pathway and network architecture in Gram-positive bacteria.
Keywords: Bacillus subtilis; Genome-wide screening; auxotrophic gene; competence; essential gene; growth; high-throughput methods for transformation and double-mutant analysis; ordered gene deletion mutant library; sporulation; ysaA.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
