

Who's Who? Detecting and Resolving Sample Anomalies in Human DNA Sequencing Studies with Peddy
- PMID: 28190455
- PMCID: PMC5339084
- DOI: 10.1016/j.ajhg.2017.01.017
Who's Who? Detecting and Resolving Sample Anomalies in Human DNA Sequencing Studies with Peddy
Abstract
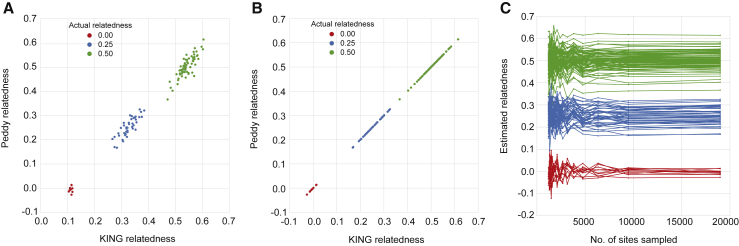
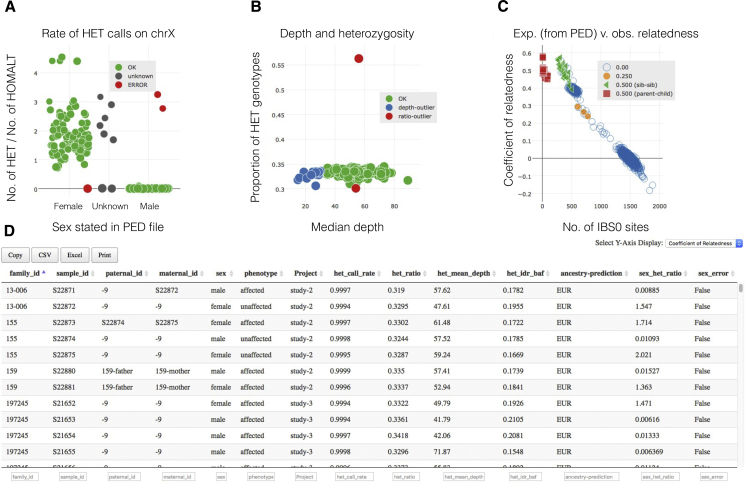
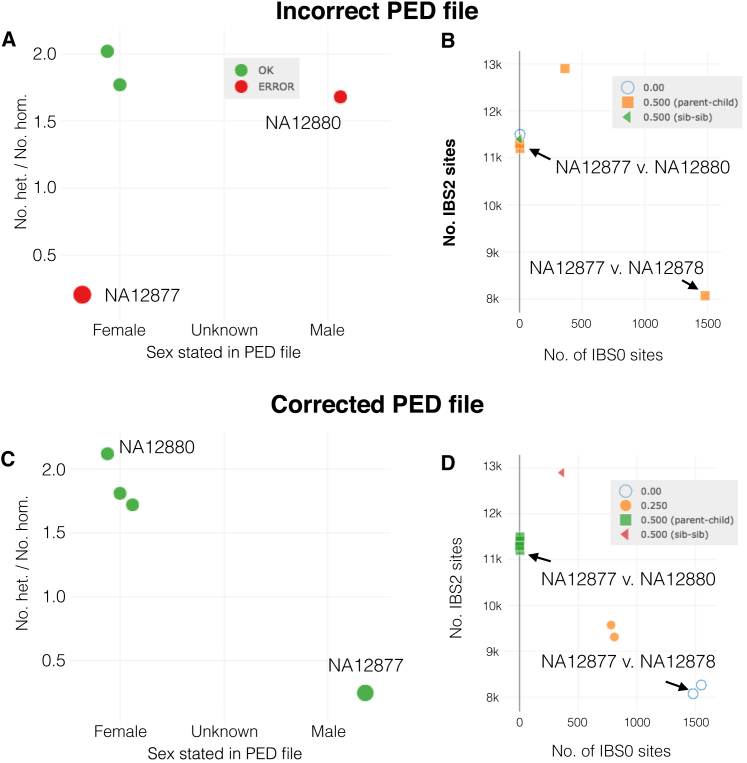
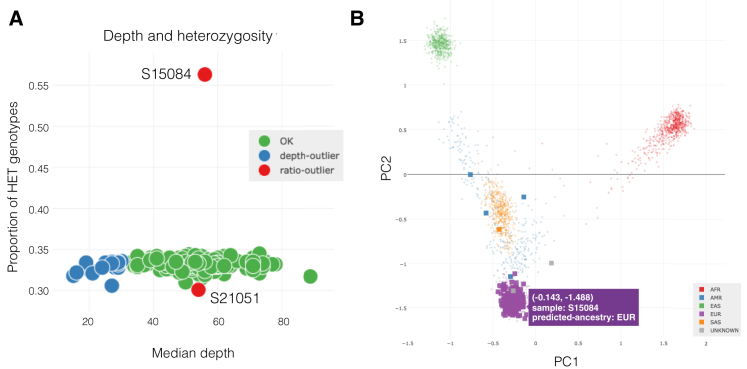
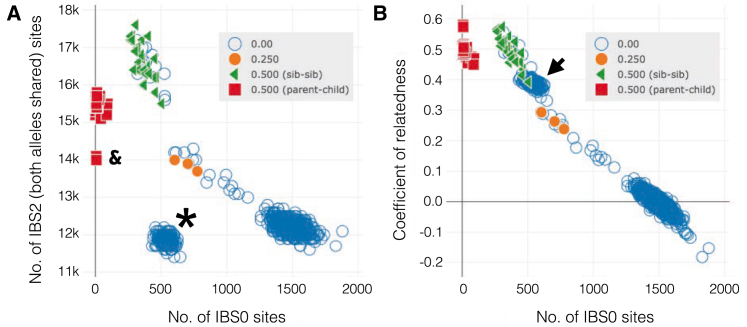
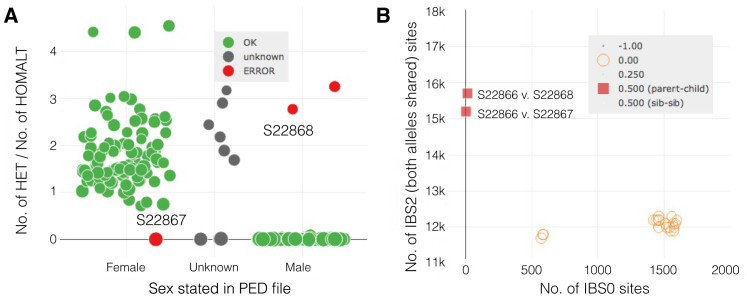
The potential for genetic discovery in human DNA sequencing studies is greatly diminished if DNA samples from a cohort are mislabeled, swapped, or contaminated or if they include unintended individuals. Unfortunately, the potential for such errors is significant since DNA samples are often manipulated by several protocols, labs, or scientists in the process of sequencing. We have developed a software package, peddy, to identify and facilitate the remediation of such errors via interactive visualizations and reports comparing the stated sex, relatedness, and ancestry to what is inferred from the individual genotypes derived from whole-genome (WGS) or whole-exome (WES) sequencing. Peddy predicts a sample's ancestry using a machine learning model trained on individuals of diverse ancestries from the 1000 Genomes Project reference panel. Peddy facilitates both automated and interactive, visual detection of sample swaps, poor sequencing quality, and other indicators of sample problems that, if left undetected, would inhibit discovery.
Keywords: QC; VCF; genetic variation; pedigree; quality control; sample mixup.
Copyright © 2017 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
