

The evolution of tumour phylogenetics: principles and practice
- PMID: 28190876
- PMCID: PMC5886015
- DOI: 10.1038/nrg.2016.170
The evolution of tumour phylogenetics: principles and practice
Abstract
Rapid advances in high-throughput sequencing and a growing realization of the importance of evolutionary theory to cancer genomics have led to a proliferation of phylogenetic studies of tumour progression. These studies have yielded not only new insights but also a plethora of experimental approaches, sometimes reaching conflicting or poorly supported conclusions. Here, we consider this body of work in light of the key computational principles underpinning phylogenetic inference, with the goal of providing practical guidance on the design and analysis of scientifically rigorous tumour phylogeny studies. We survey the range of methods and tools available to the researcher, their key applications, and the various unsolved problems, closing with a perspective on the prospects and broader implications of this field.
Figures
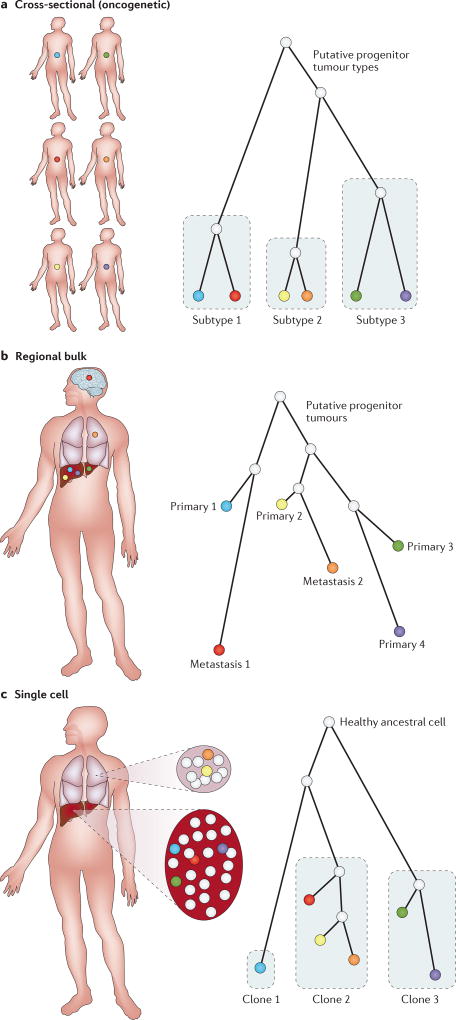
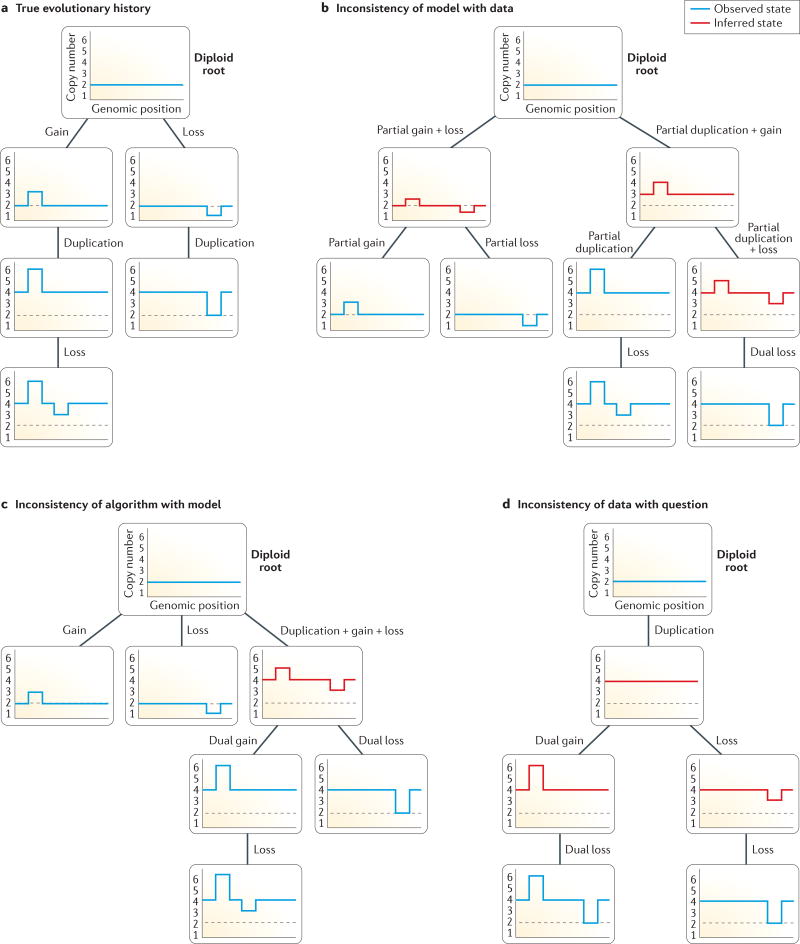
References
-
- Hanks S, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in (BUB1B) Nat. Genet. 2004;36:1159–1161. - PubMed
-
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. This is a seminal paper proposing that solid tumours evolve clonally while accumulating mutations from one mitosis to the next via a process of selection of mutant subpopulations from a common progenitor cell. - PubMed
-
- Polyak K. Is breast tumor progression really linear? Clin. Cancer Res. 2008;14:339–341. - PubMed
-
- Naxerova K, Jain RK. Using tumour phylogenetics to identify the roots of metastasis in humans. Nat. Rev. Clin. Oncol. 2015;12:258–272. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
