

Mutations in DONSON disrupt replication fork stability and cause microcephalic dwarfism
- PMID: 28191891
- PMCID: PMC5450907
- DOI: 10.1038/ng.3790
Mutations in DONSON disrupt replication fork stability and cause microcephalic dwarfism
Abstract
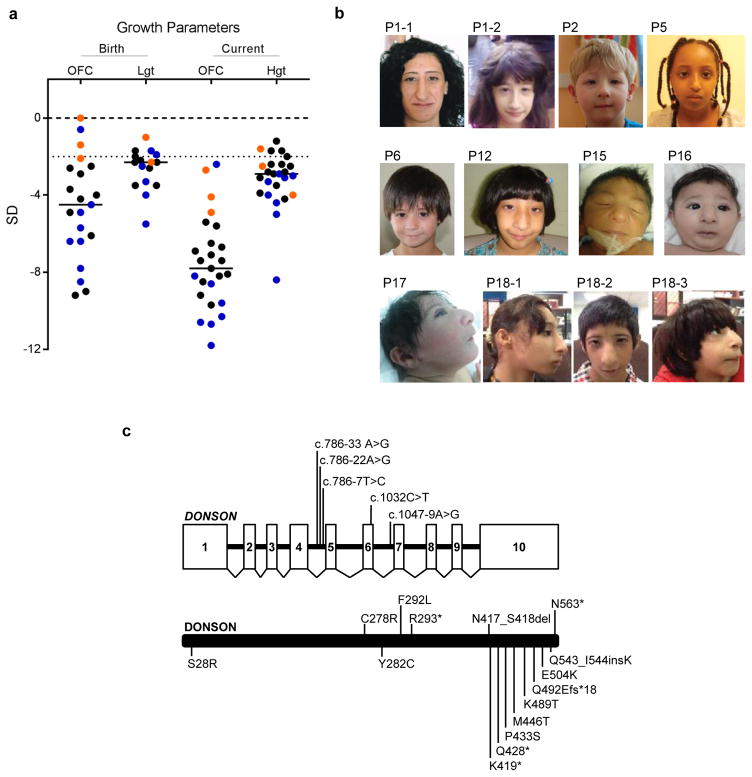
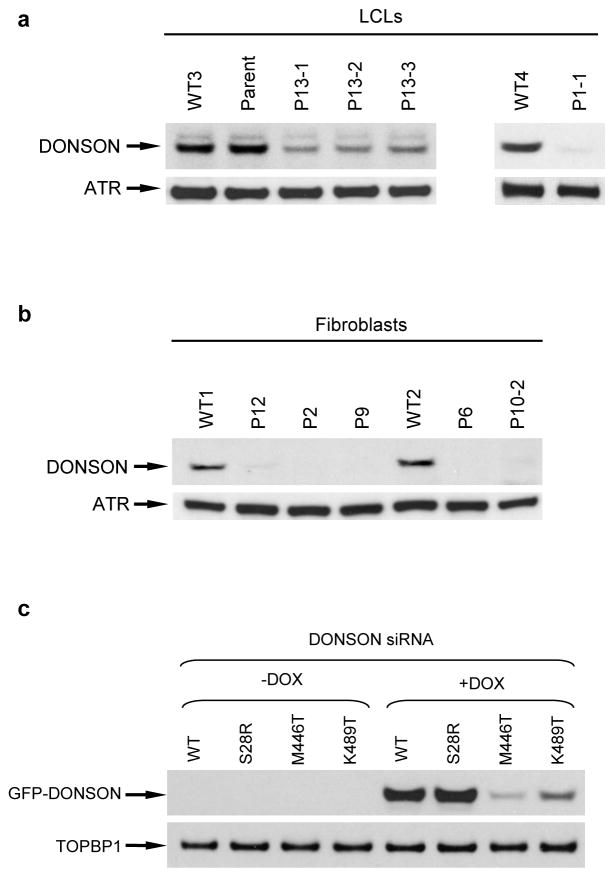
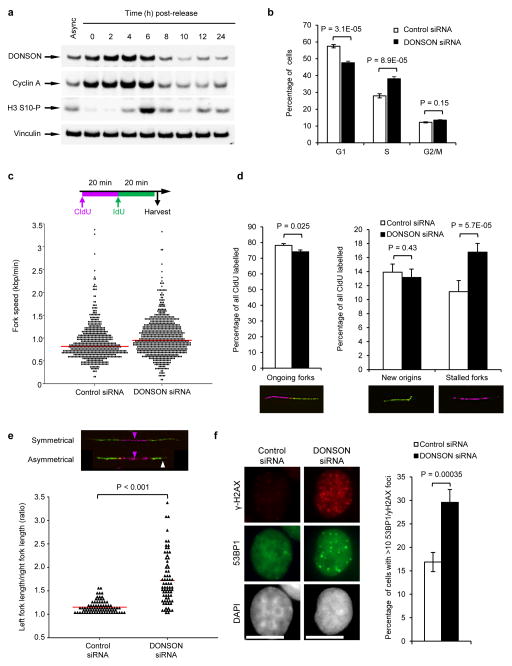
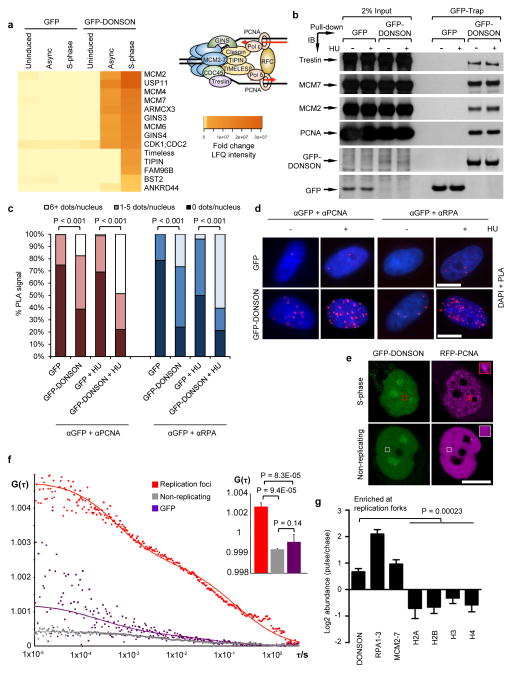
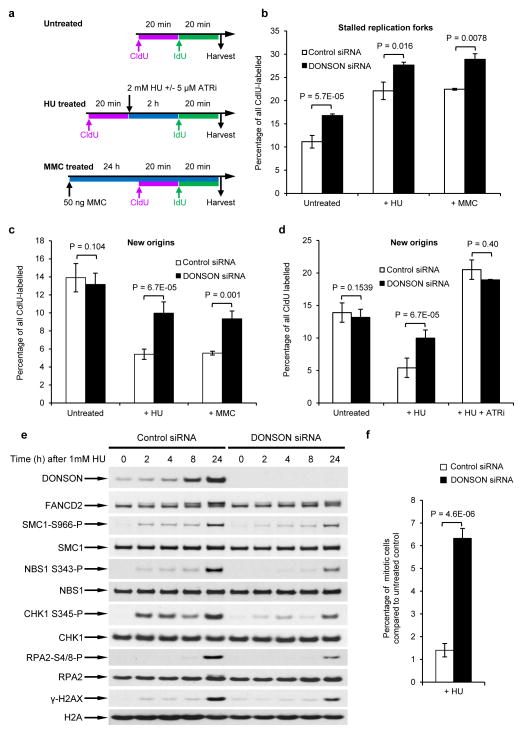
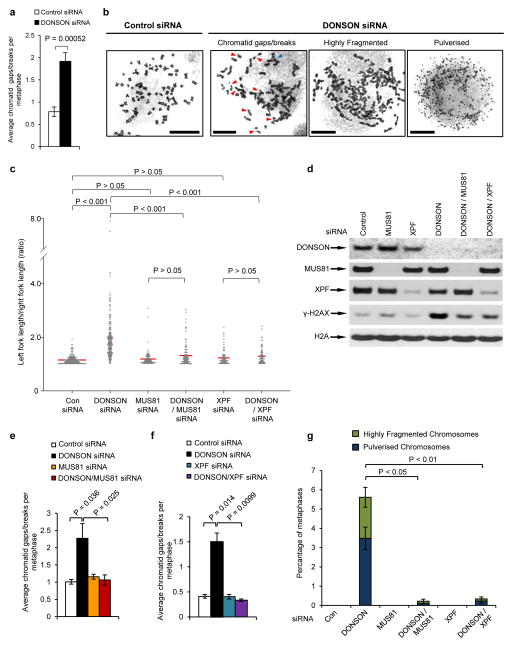
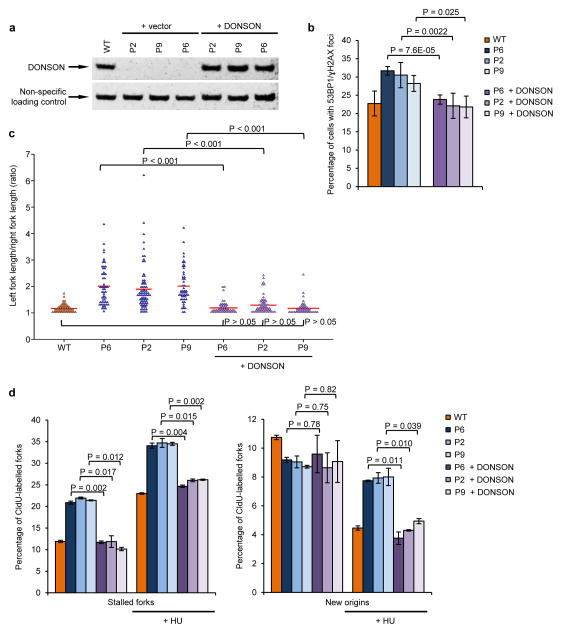
To ensure efficient genome duplication, cells have evolved numerous factors that promote unperturbed DNA replication and protect, repair and restart damaged forks. Here we identify downstream neighbor of SON (DONSON) as a novel fork protection factor and report biallelic DONSON mutations in 29 individuals with microcephalic dwarfism. We demonstrate that DONSON is a replisome component that stabilizes forks during genome replication. Loss of DONSON leads to severe replication-associated DNA damage arising from nucleolytic cleavage of stalled replication forks. Furthermore, ATM- and Rad3-related (ATR)-dependent signaling in response to replication stress is impaired in DONSON-deficient cells, resulting in decreased checkpoint activity and the potentiation of chromosomal instability. Hypomorphic mutations in DONSON substantially reduce DONSON protein levels and impair fork stability in cells from patients, consistent with defective DNA replication underlying the disease phenotype. In summary, we have identified mutations in DONSON as a common cause of microcephalic dwarfism and established DONSON as a critical replication fork protein required for mammalian DNA replication and genome stability.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
