

Nuclear genetic codes with a different meaning of the UAG and the UAA codon
- PMID: 28193262
- PMCID: PMC5304391
- DOI: 10.1186/s12915-017-0353-y
Nuclear genetic codes with a different meaning of the UAG and the UAA codon
Abstract
Background: Departures from the standard genetic code in eukaryotic nuclear genomes are known for only a handful of lineages and only a few genetic code variants seem to exist outside the ciliates, the most creative group in this regard. Most frequent code modifications entail reassignment of the UAG and UAA codons, with evidence for at least 13 independent cases of a coordinated change in the meaning of both codons. However, no change affecting each of the two codons separately has been documented, suggesting the existence of underlying evolutionary or mechanistic constraints.
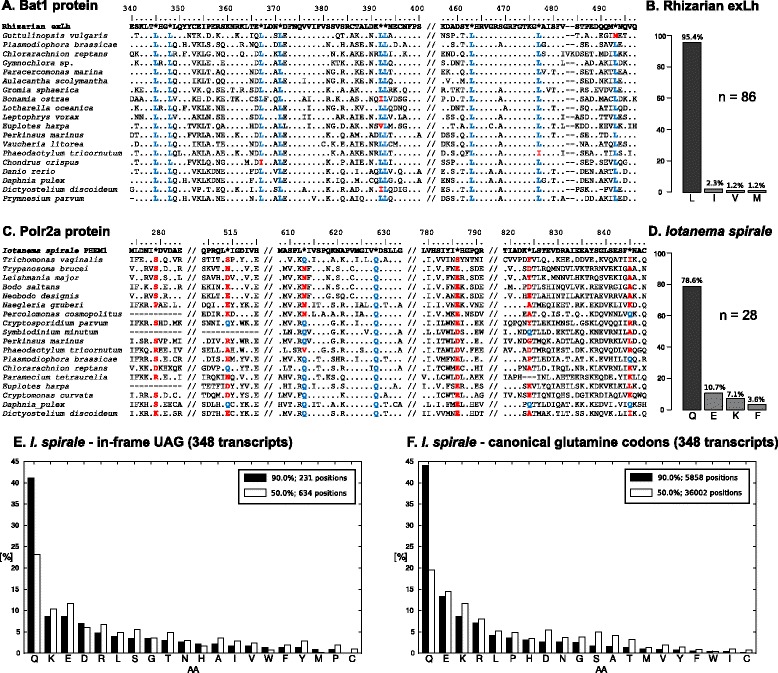
Results: Here, we present the discovery of two new variants of the nuclear genetic code, in which UAG is translated as an amino acid while UAA is kept as a termination codon (along with UGA). The first variant occurs in an organism noticed in a (meta)transcriptome from the heteropteran Lygus hesperus and demonstrated to be a novel insect-dwelling member of Rhizaria (specifically Sainouroidea). This first documented case of a rhizarian with a non-canonical genetic code employs UAG to encode leucine and represents an unprecedented change among nuclear codon reassignments. The second code variant was found in the recently described anaerobic flagellate Iotanema spirale (Metamonada: Fornicata). Analyses of transcriptomic data revealed that I. spirale uses UAG to encode glutamine, similarly to the most common variant of a non-canonical code known from several unrelated eukaryotic groups, including hexamitin diplomonads (also a lineage of fornicates). However, in these organisms, UAA also encodes glutamine, whereas it is the primary termination codon in I. spirale. Along with phylogenetic evidence for distant relationship of I. spirale and hexamitins, this indicates two independent genetic code changes in fornicates.
Conclusions: Our study documents, for the first time, that evolutionary changes of the meaning of UAG and UAA codons in nuclear genomes can be decoupled and that the interpretation of the two codons by the cytoplasmic translation apparatus is mechanistically separable. The latter conclusion has interesting implications for possibilities of genetic code engineering in eukaryotes. We also present a newly developed generally applicable phylogeny-informed method for inferring the meaning of reassigned codons.
Keywords: Codon reassignment; Evolution; Evolutionary constraint; Fornicata; Genetic code; Iotanema spirale; Lygus hesperus; Protists; Rhizaria; Transcriptome.
Figures



References
-
- Matsumoto T, Ishikawa SA, Hashimoto T, Inagaki Y. A deviant genetic code in the green alga-derived plastid in the dinoflagellate Lepidodinium chlorophorum. Mol Phylogenet Evol. 2011;60:68–72. - PubMed
-
- Preer Jr JR, Preer LB, Rudman BM, Barnett AJ. Deviation from the universal code shown by the gene for surface protein 51A in Paramecium. Nature. 1985;314:188–90. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
