

Towards Selective Mycobacterial ClpP1P2 Inhibitors with Reduced Activity against the Human Proteasome
- PMID: 28193668
- PMCID: PMC5404560
- DOI: 10.1128/AAC.02307-16
Towards Selective Mycobacterial ClpP1P2 Inhibitors with Reduced Activity against the Human Proteasome
Abstract
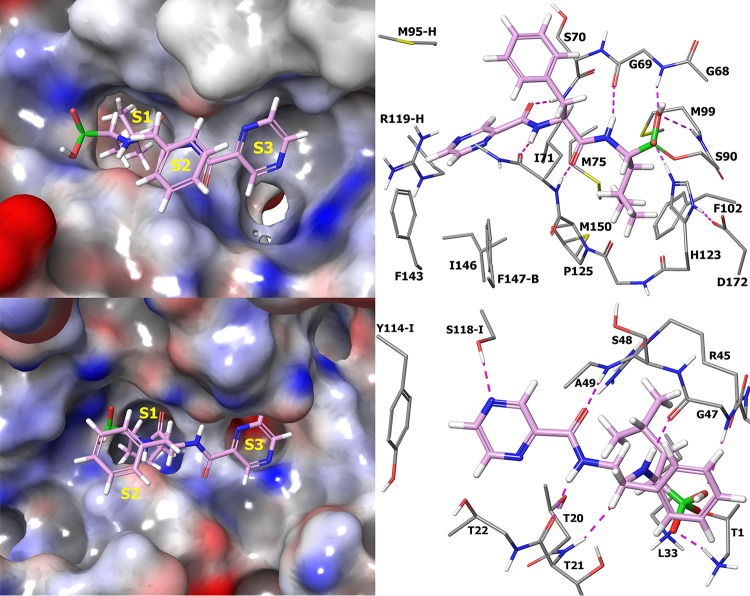
Mycobacterium tuberculosis is responsible for the greatest number of deaths worldwide due to a bacterial agent. We recently identified bortezomib (Velcade; compound 1) as a promising antituberculosis (anti-TB) compound. We showed that compound 1 inhibits the mycobacterial caseinolytic proteases P1 and P2 (ClpP1P2) and exhibits bactericidal activity, and we established compound 1 and ClpP1P2 as an attractive lead/target couple. However, compound 1 is a human-proteasome inhibitor currently approved for cancer therapy and, as such, exhibits significant toxicity. Selective inhibition of the bacterial protease over the human proteasome is desirable in order to maintain antibacterial activity while reducing toxicity. We made use of structural data in order to design a series of dipeptidyl-boronate derivatives of compound 1. We tested these derivatives for whole-cell ClpP1P2 and human-proteasome inhibition as well as bacterial-growth inhibition and identified compounds that were up to 100-fold-less active against the human proteasome but that retained ClpP1P2 and mycobacterial-growth inhibition as well as bactericidal potency. The lead compound, compound 58, had low micromolar ClpP1P2 and anti-M. tuberculosis activity, good aqueous solubility, no cytochrome P450 liabilities, moderate plasma protein binding, and low toxicity in two human liver cell lines, and despite high clearance in microsomes, this compound was only moderately cleared when administered intravenously or orally to mice. Higher-dose oral pharmacokinetics indicated good dose linearity. Furthermore, compound 58 was inhibitory to only 11% of a panel of 62 proteases. Our work suggests that selectivity over the human proteasome can be achieved with a drug-like template while retaining potency against ClpP1P2 and, crucially, anti-M. tuberculosis activity.
Keywords: antimycobacterial; caseinolytic protease; dipeptidyl boronic acid.
Copyright © 2017 Moreira et al.
Figures









References
-
- WHO. 2016. Global tuberculosis report 2016. WHO, Geneva, Switzerland.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
