

Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington's disease
- PMID: 28194040
- PMCID: PMC5316841
- DOI: 10.1038/ncomms14405
Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington's disease
Abstract
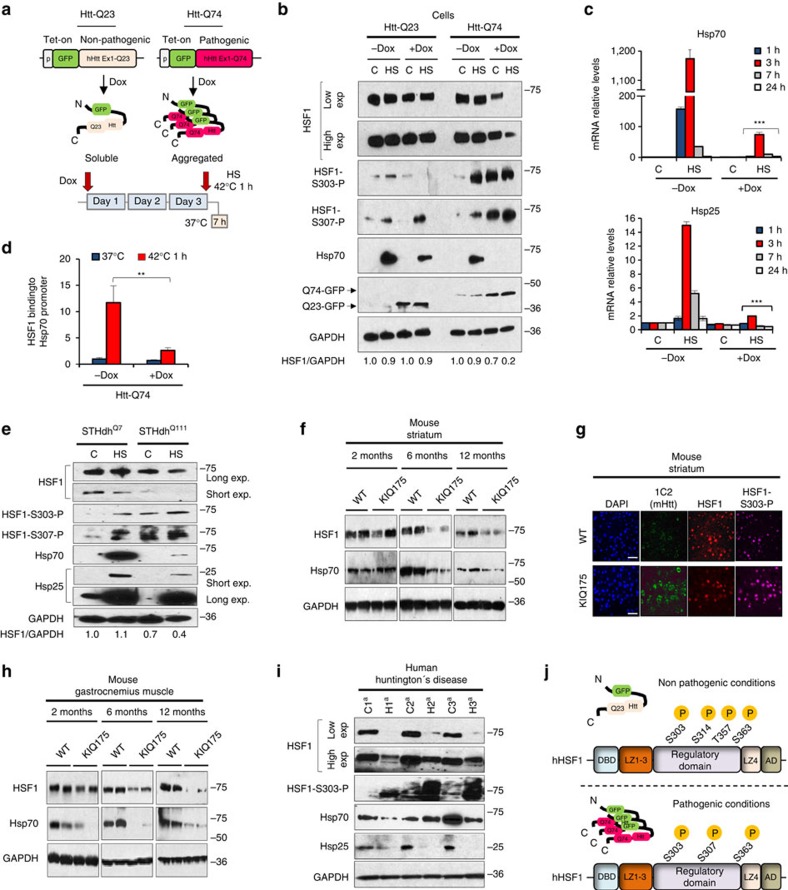
Huntington's Disease (HD) is a neurodegenerative disease caused by poly-glutamine expansion in the Htt protein, resulting in Htt misfolding and cell death. Expression of the cellular protein folding and pro-survival machinery by heat shock transcription factor 1 (HSF1) ameliorates biochemical and neurobiological defects caused by protein misfolding. We report that HSF1 is degraded in cells and mice expressing mutant Htt, in medium spiny neurons derived from human HD iPSCs and in brain samples from patients with HD. Mutant Htt increases CK2α' kinase and Fbxw7 E3 ligase levels, phosphorylating HSF1 and promoting its proteasomal degradation. An HD mouse model heterozygous for CK2α' shows increased HSF1 and chaperone levels, maintenance of striatal excitatory synapses, clearance of Htt aggregates and preserves body mass compared with HD mice homozygous for CK2α'. These results reveal a pathway that could be modulated to prevent neuronal dysfunction and muscle wasting caused by protein misfolding in HD.
Conflict of interest statement
D.J.T. is a founder of Chaperone Therapeutics and a member of the SAB.
Figures








References
-
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72, 971–983 (1993). - PubMed
-
- Novak M. J. & Tabrizi S. J. Huntington's disease. BMJ 340, c3109 (2010). - PubMed
-
- Saudou F. & Humbert S. The biology of huntingtin. Neuron 89, 910–926 (2016). - PubMed
-
- Sassone J., Colciago C., Cislaghi G., Silani V. & Ciammola A. Huntington's disease: the current state of research with peripheral tissues. Exp. Neurol. 219, 385–397 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
