

R248G cystic fibrosis transmembrane conductance regulator mutation in three siblings presenting with recurrent acute pancreatitis and reproductive issues: a case series
- PMID: 28196530
- PMCID: PMC5310058
- DOI: 10.1186/s13256-016-1181-3
R248G cystic fibrosis transmembrane conductance regulator mutation in three siblings presenting with recurrent acute pancreatitis and reproductive issues: a case series
Abstract
Background: Mutational combinations of the cystic fibrosis transmembrane conductance regulator, CFTR, gene have different phenotypic manifestations at the molecular level with varying clinical consequences for individuals possessing such mutations. Reporting cystic fibrosis transmembrane conductance regulator mutations is important in understanding the genotype-phenotype correlations and associated clinical presentations in patients with cystic fibrosis. Understanding the effects of mutations is critical in developing appropriate treatments for individuals affected with cystic fibrosis, non-classic cystic fibrosis, or cystic fibrosis transmembrane conductance regulator-related disorders. This is the first report of related individuals possessing the R248G missense cystic fibrosis transmembrane conductance regulator mutation and we present their associated clinical histories.
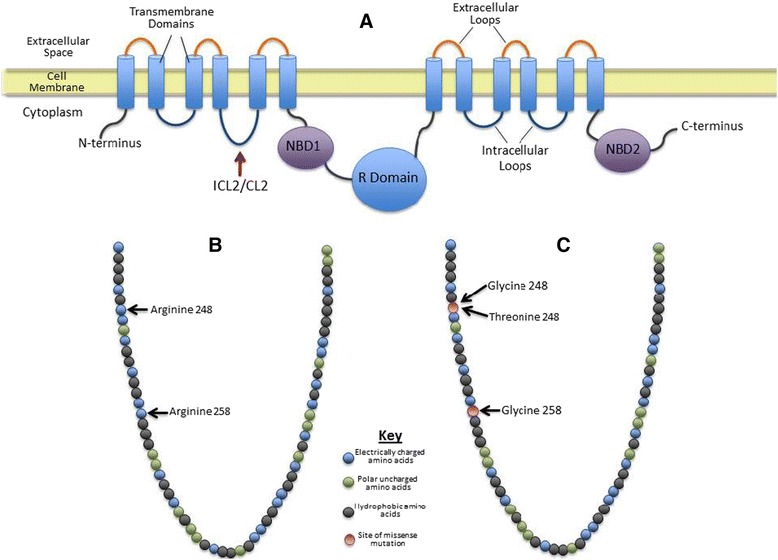
Case presentation: All three patients are of Spanish descent. Deoxyribonucleic acid analysis revealed that all three siblings possessed a novel c.742A>G mutation, resulting in a p.Arg248Gly (R248G) amino acid change in exon 6 in trans with the known N1303K mutant allele. Case 1 patient is a 39-year-old infertile man presenting with congenital unilateral absence of the vas deferens and recurrent episodes of epigastric pain. Case 2 patient is a 32-year-old woman presenting with periods of infertility, two previous spontaneous abortions, recurrent epigastric pain, and recurrent pancreatitis. Case 3 patient is a 29-year-old woman presenting with recurrent pancreatitis and epigastric pain.
Conclusions: We report the genotype-phenotype correlations and clinical manifestations of a novel R248G cystic fibrosis transmembrane conductance regulator mutation: congenital unilateral absence of the vas deferens in males, reduced female fertility, and recurrent acute pancreatitis. In addition, we discuss the possible functional consequences of the mutations at the molecular level.
Keywords: CFTR; Congenital absence of vas deferens; Genotype-phenotype; Missense mutation.
Figures
References
-
- Bombieri C, Claustres M, De Boeck K, Derichs N, Dodge J, Girodon E, Sermet I, Schwarz M, Tzetis M, Wilschanski M, Bareil C, Bilton D, et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros. 2011;10:86–102. doi: 10.1016/S1569-1993(11)60014-3. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
