

Identification of a transcriptional activation domain in yeast repressor activator protein 1 (Rap1) using an altered DNA-binding specificity variant
- PMID: 28196871
- PMCID: PMC5392566
- DOI: 10.1074/jbc.M117.779181
Identification of a transcriptional activation domain in yeast repressor activator protein 1 (Rap1) using an altered DNA-binding specificity variant
Abstract
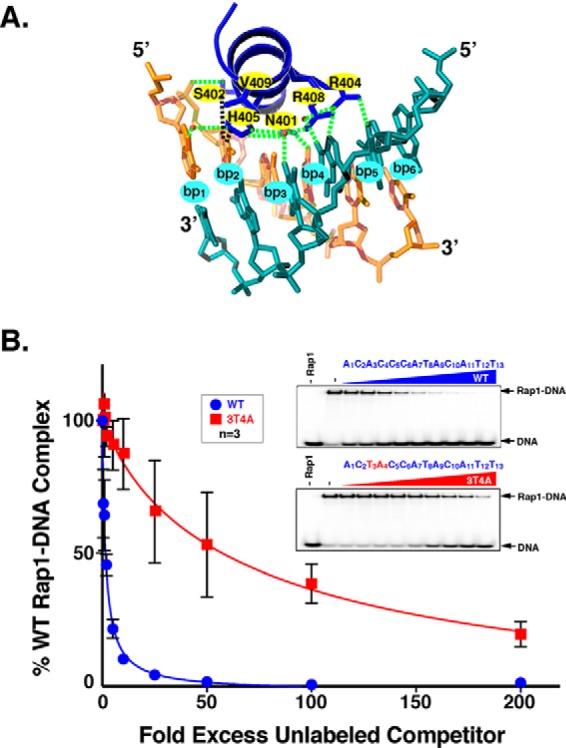
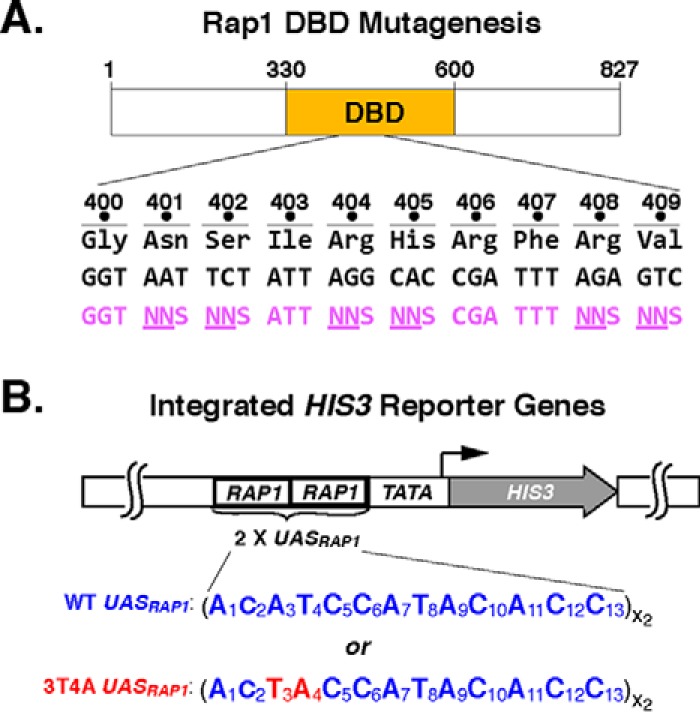
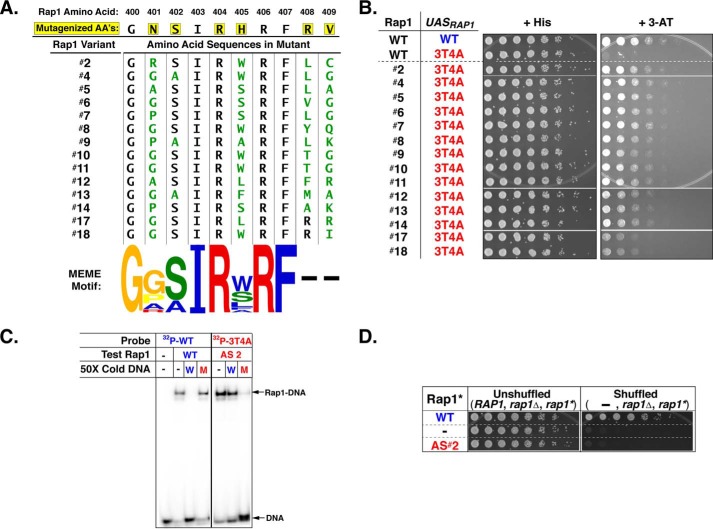
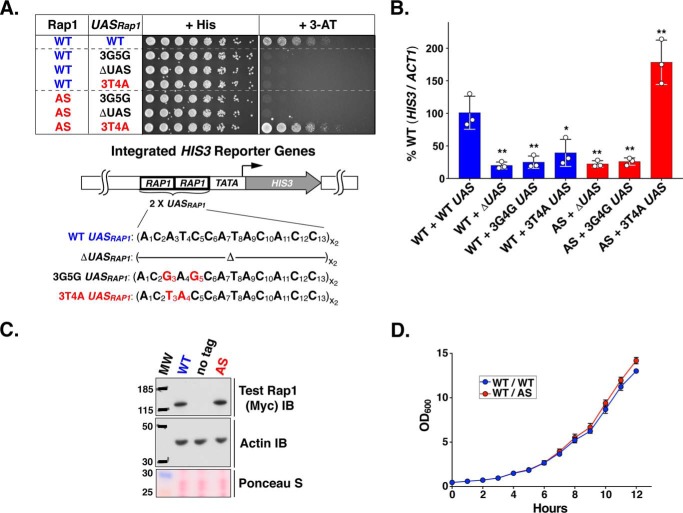
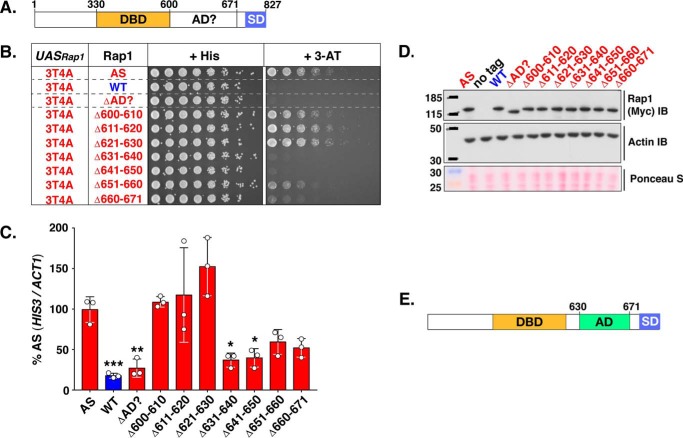
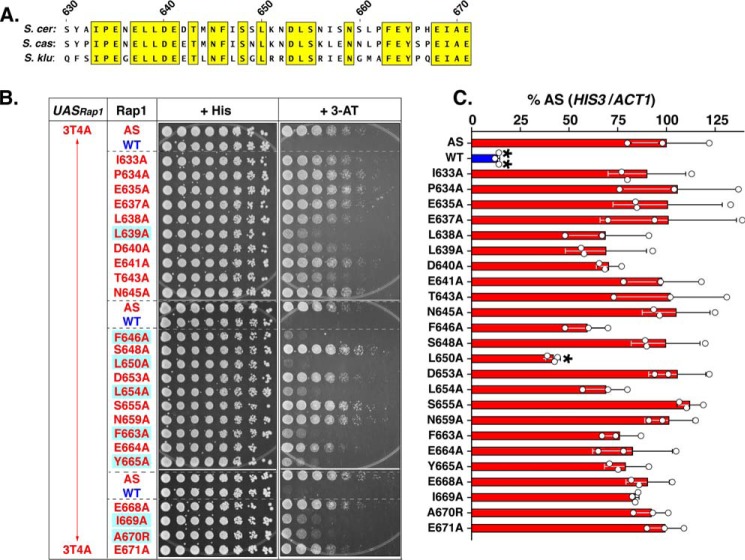
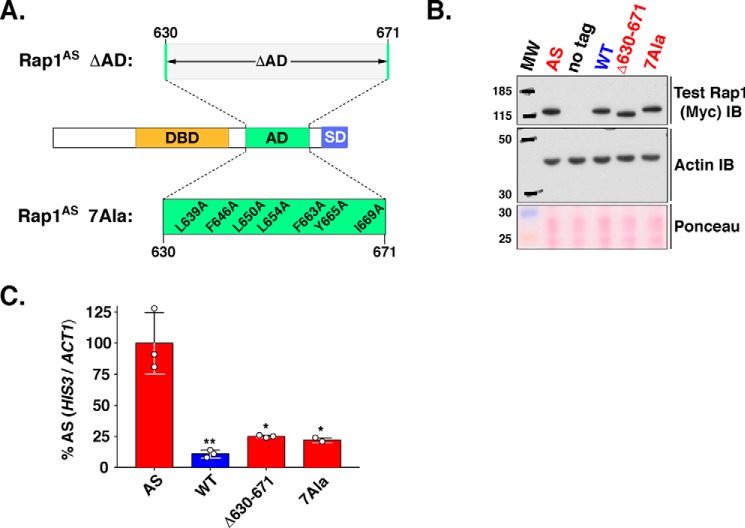
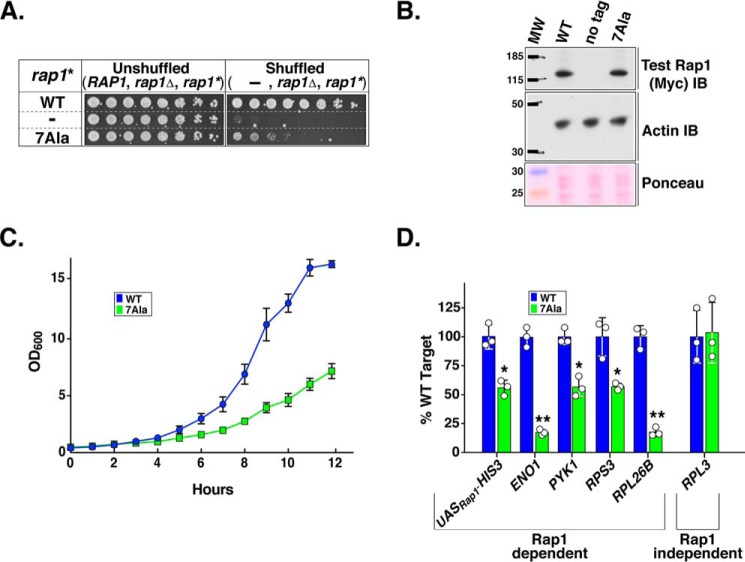
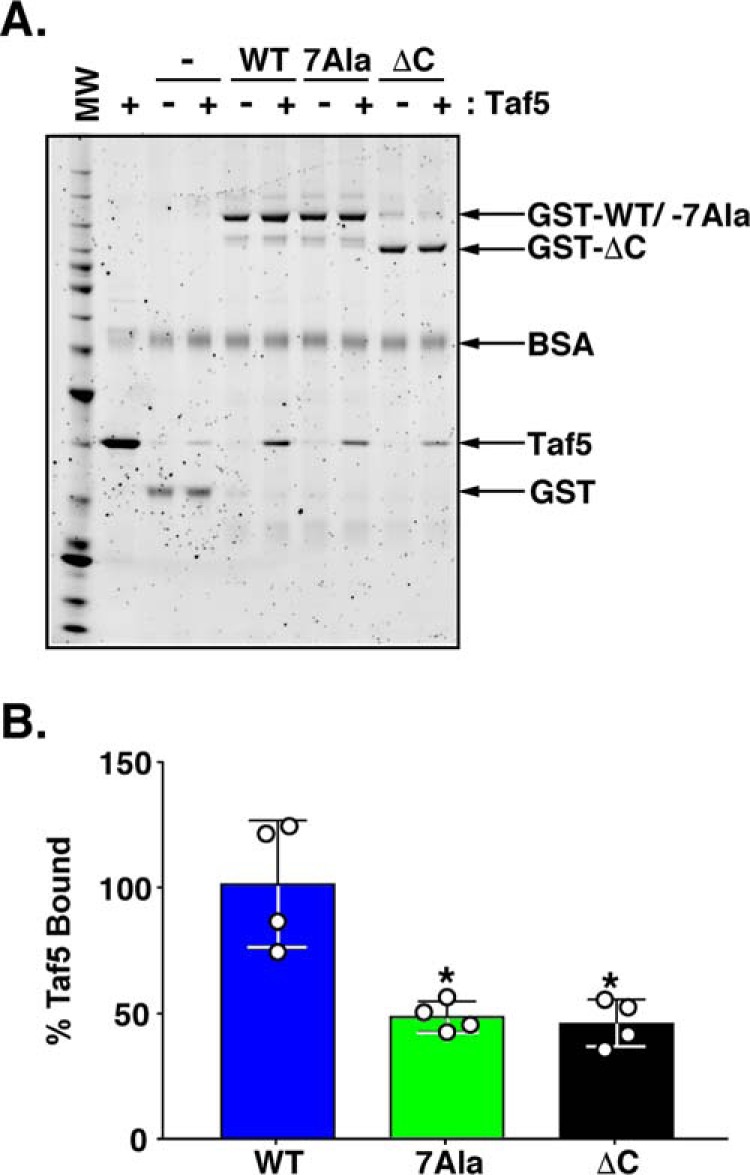
Repressor activator protein 1 (Rap1) performs multiple vital cellular functions in the budding yeast Saccharomyces cerevisiae These include regulation of telomere length, transcriptional repression of both telomere-proximal genes and the silent mating type loci, and transcriptional activation of hundreds of mRNA-encoding genes, including the highly transcribed ribosomal protein- and glycolytic enzyme-encoding genes. Studies of the contributions of Rap1 to telomere length regulation and transcriptional repression have yielded significant mechanistic insights. However, the mechanism of Rap1 transcriptional activation remains poorly understood because Rap1 is encoded by a single copy essential gene and is involved in many disparate and essential cellular functions, preventing easy interpretation of attempts to directly dissect Rap1 structure-function relationships. Moreover, conflicting reports on the ability of Rap1-heterologous DNA-binding domain fusion proteins to serve as chimeric transcriptional activators challenge use of this approach to study Rap1. Described here is the development of an altered DNA-binding specificity variant of Rap1 (Rap1AS). We used Rap1AS to map and characterize a 41-amino acid activation domain (AD) within the Rap1 C terminus. We found that this AD is required for transcription of both chimeric reporter genes and authentic chromosomal Rap1 enhancer-containing target genes. Finally, as predicted for a bona fide AD, mutation of this newly identified AD reduced the efficiency of Rap1 binding to a known transcriptional coactivator TFIID-binding target, Taf5. In summary, we show here that Rap1 contains an AD required for Rap1-dependent gene transcription. The Rap1AS variant will likely also be useful for studies of the functions of Rap1 in other biological pathways.
Keywords: Saccharomyces cerevisiae; altered DNA binding specificity; gene regulation; protein engineering; transcription activation; transcription activator; transcription regulation; yeast.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Taatjes D. J., Marr M. T., and Tjian R. (2004) Regulatory diversity among metazoan co-activator complexes. Nat. Rev. Mol. Cell Biol. 5, 403–410 - PubMed
-
- Shandilya J., and Roberts S. G. (2012) The transcription cycle in eukaryotes: from productive initiation to RNA polymerase II recycling. Biochim. Biophys. Acta 1819, 391–400 - PubMed
-
- Weake V. M., and Workman J. L. (2010) Inducible gene expression: diverse regulatory mechanisms. Nat. Rev. Genet. 11, 426–437 - PubMed
-
- Ranish J. A., and Hahn S. (1996) Transcription: basal factors and activation. Curr. Opin. Genet. Dev. 6, 151–158 - PubMed
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
