

Enhancing genomic prediction with genome-wide association studies in multiparental maize populations
- PMID: 28198815
- PMCID: PMC5436027
- DOI: 10.1038/hdy.2017.4
Enhancing genomic prediction with genome-wide association studies in multiparental maize populations
Abstract
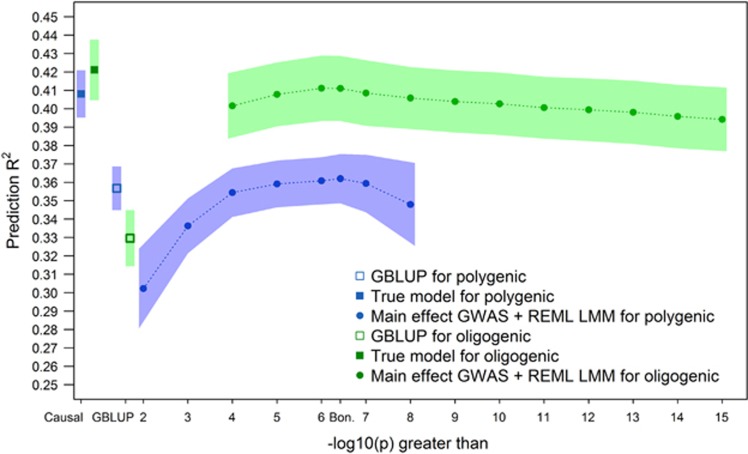
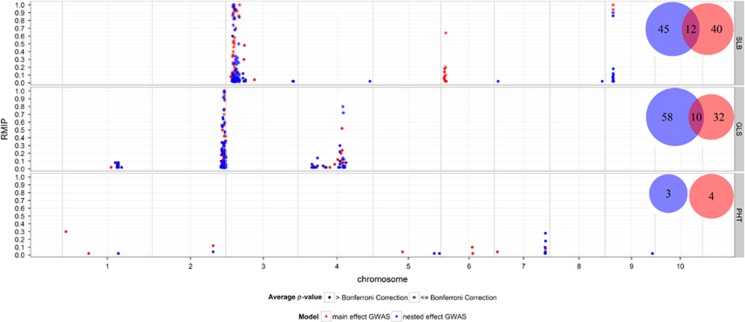
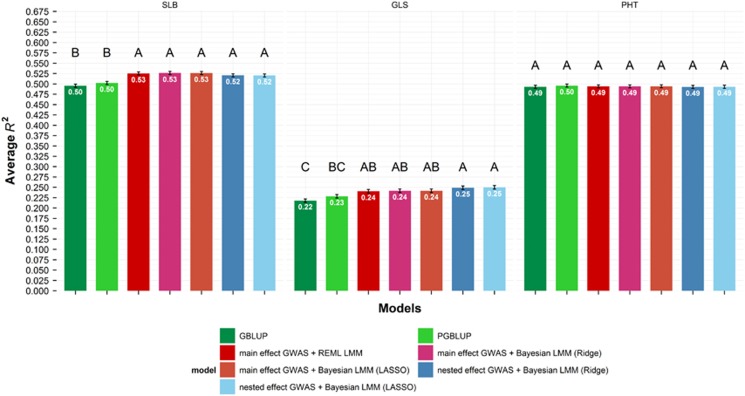
Genome-wide association mapping using dense marker sets has identified some nucleotide variants affecting complex traits that have been validated with fine-mapping and functional analysis. However, many sequence variants associated with complex traits in maize have small effects and low repeatability. In contrast to genome-wide association study (GWAS), genomic prediction (GP) is typically based on models incorporating information from all available markers, rather than modeling effects of individual loci. We considered methods to integrate results of GWASs into GP models in the context of multiple interconnected families. We compared association tests based on a biallelic additive model constraining the effect of a single-nucleotide polymorphism (SNP) to be equal across all families in which it segregates to a model in which the effect of a SNP can vary across families. Association SNPs were then included as fixed effects into a GP model that also included the random effects of the whole genome background. Simulation studies revealed that the effectiveness of this joint approach depends on the extent of polygenicity of the traits. Congruent with this finding, cross-validation studies indicated that GP including the fixed effects of the most significantly associated SNPs along with the polygenic background was more accurate than the polygenic background model alone for moderately complex but not highly polygenic traits measured in the maize nested association mapping population. Individual SNPs with strong and robust association signals can effectively improve GP. Our approach provides a new integrative modeling approach for both reliable gene discovery and robust GP.
Figures
References
-
- Beavis WD. (1998). QTL analyses: power, precision, and accuracy. In: Paterson AJ (ed). Molecular Dissection of Complex Traits. CRC Press: Boca Raton, FL, USA, pp 145–162.
-
- Bernardo R. (2001). What if we knew all the genes for a quantitative trait in hybrid crops? Crop Sci 41: 1–4.
-
- Bernardo R. (2014). Genomewide selection when major genes are known. Crop Sci 54: 68–75.
-
- Bernardo R, Yu J. (2007). Prospects for genomewide selection for quantitative traits in maize. Crop Sci 47: 1082–1090.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
