

Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms
- PMID: 28202457
- PMCID: PMC5399482
- DOI: 10.1182/blood-2016-10-743302
Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms
Abstract
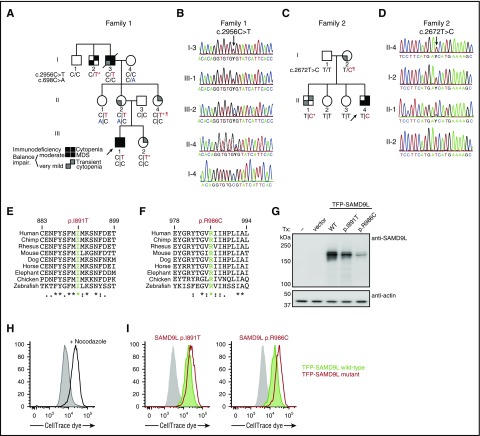
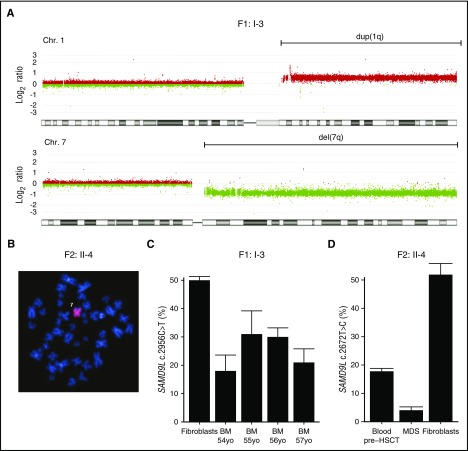
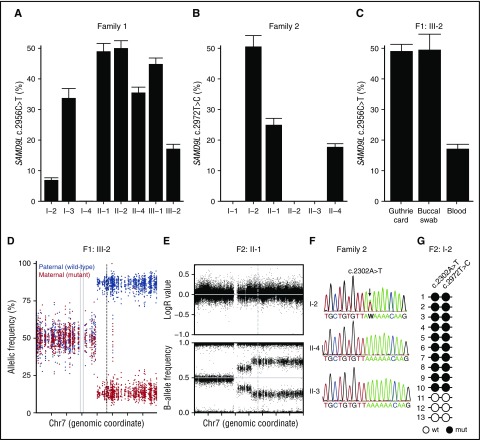
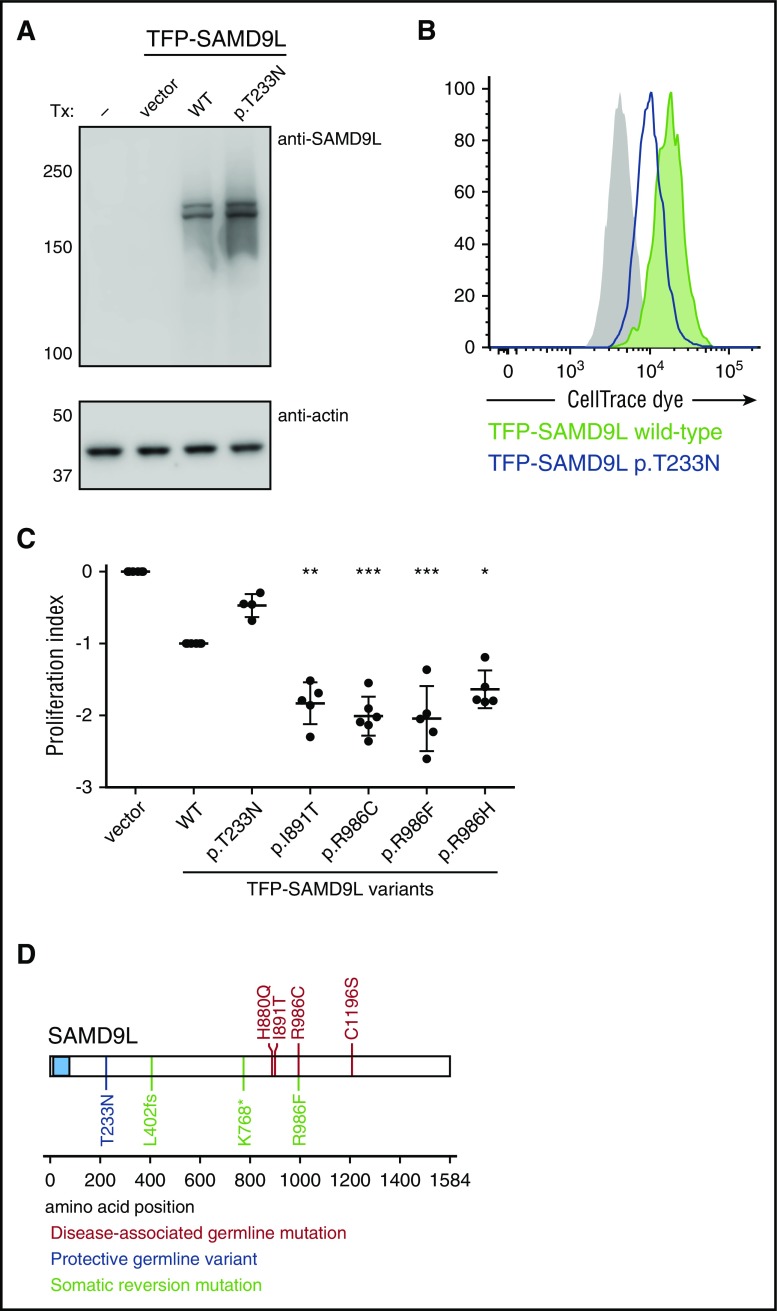
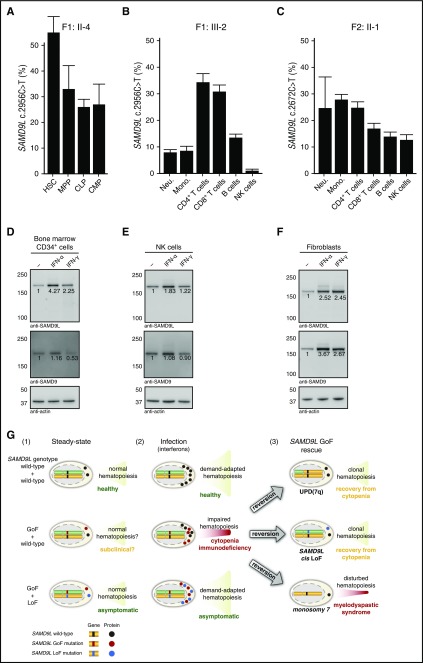
Several monogenic causes of familial myelodysplastic syndrome (MDS) have recently been identified. We studied 2 families with cytopenia, predisposition to MDS with chromosome 7 aberrations, immunodeficiency, and progressive cerebellar dysfunction. Genetic studies uncovered heterozygous missense mutations in SAMD9L, a tumor suppressor gene located on chromosome arm 7q. Consistent with a gain-of-function effect, ectopic expression of the 2 identified SAMD9L mutants decreased cell proliferation relative to wild-type protein. Of the 10 individuals identified who were heterozygous for either SAMD9L mutation, 3 developed MDS upon loss of the mutated SAMD9L allele following intracellular infections associated with myeloid, B-, and natural killer (NK)-cell deficiency. Five other individuals, 3 with spontaneously resolved cytopenic episodes in infancy, harbored hematopoietic revertant mosaicism by uniparental disomy of 7q, with loss of the mutated allele or additional in cisSAMD9L truncating mutations. Examination of 1 individual indicated that somatic reversions were postnatally selected. Somatic mutations were tracked to CD34+ hematopoietic progenitor cell populations, being further enriched in B and NK cells. Stimulation of these cell types with interferon (IFN)-α or IFN-γ induced SAMD9L expression. Clinically, revertant mosaicism was associated with milder disease, yet neurological manifestations persisted in 3 individuals. Two carriers also harbored a rare, in trans germ line SAMD9L missense loss-of-function variant, potentially counteracting the SAMD9L mutation. Our results demonstrate that gain-of-function mutations in the tumor suppressor SAMD9L cause cytopenia, immunodeficiency, variable neurological presentation, and predisposition to MDS with -7/del(7q), whereas hematopoietic revertant mosaicism commonly ameliorated clinical manifestations. The findings suggest a role for SAMD9L in regulating IFN-driven, demand-adapted hematopoiesis.
© 2017 by The American Society of Hematology.
Figures

Comment in
-
I am SAMD9L: 7q regulator I am.Blood. 2017 Apr 20;129(16):2210-2212. doi: 10.1182/blood-2017-03-770198. Blood. 2017. PMID: 28428236 No abstract available.
References
-
- Arber DA, Orazi A, Hasserjian R, et al. . The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391-2405. - PubMed
-
- Adès L, Itzykson R, Fenaux P. Myelodysplastic syndromes. Lancet. 2014;383(9936):2239-2252. - PubMed
-
- Haase D, Germing U, Schanz J, et al. . New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110(13):4385-4395. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
