

Clinical and genetic factors predicting Dravet syndrome in infants with SCN1A mutations
- PMID: 28202706
- PMCID: PMC5384833
- DOI: 10.1212/WNL.0000000000003716
Clinical and genetic factors predicting Dravet syndrome in infants with SCN1A mutations
Abstract
Objective: To explore the prognostic value of initial clinical and mutational findings in infants with SCN1A mutations.
Methods: Combining sex, age/fever at first seizure, family history of epilepsy, EEG, and mutation type, we analyzed the accuracy of significant associations in predicting Dravet syndrome vs milder outcomes in 182 mutation carriers ascertained after seizure onset. To assess the diagnostic accuracy of all parameters, we calculated sensitivity, specificity, receiver operating characteristic (ROC) curves, diagnostic odds ratios, and positive and negative predictive values and the accuracy of combined information. We also included in the study demographic and mutational data of the healthy relatives of mutation carrier patients.
Results: Ninety-seven individuals (48.5%) had Dravet syndrome, 49 (23.8%) had generalized/genetic epilepsy with febrile seizures plus, 30 (14.8%) had febrile seizures, 6 (3.5%) had focal epilepsy, and 18 (8.9%) were healthy relatives. The association study indicated that age at first seizure and frameshift mutations were associated with Dravet syndrome. The risk of Dravet syndrome was 85% in the 0- to 6-month group, 51% in the 6- to 12-month range, and 0% after the 12th month. ROC analysis identified onset within the sixth month as the diagnostic cutoff for progression to Dravet syndrome (sensitivity = 83.3%, specificity = 76.6%).
Conclusions: In individuals with SCN1A mutations, age at seizure onset appears to predict outcome better than mutation type. Because outcome is not predetermined by genetic factors only, early recognition and treatment that mitigates prolonged/repeated seizures in the first year of life might also limit the progression to epileptic encephalopathy.
© 2017 American Academy of Neurology.
Figures

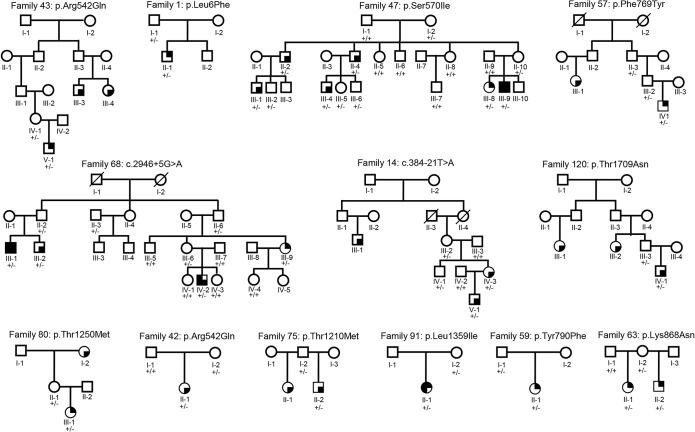
 = Dravet syndrome;
= Dravet syndrome;  = FS;
= FS;  = generalized/genetic epilepsy with febrile seizures plus;
= generalized/genetic epilepsy with febrile seizures plus;  = focal epilepsy; +/− = heterozygous SCN1A mutation; +/+ = absence of the SCN1A mutation.
= focal epilepsy; +/− = heterozygous SCN1A mutation; +/+ = absence of the SCN1A mutation.References
-
- Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA. SCN1A mutations and epilepsy. Hum Mutat 2005;25:535–542. - PubMed
-
- Guerrini R, Striano P. Dravet syndrome: not just epilepsy. Neurology 2016;87:245–246. - PubMed
-
- Guerrini R, Dravet C. Severe epileptic encephalopathies of infancy, other than West syndrome. In: Engel J, Pedley TA, editors. Epilepsy: A Comprehensive Textbook. Vol 3. Philadelphia: Lippincott; 1997:2285–2302.
-
- Hirose S, Scheffer IE, Marini C, et al. ; Genetics Commission of the International League Against Epilepsy. SCN1A testing for epilepsy: application in clinical practice. Epilepsia 2013;54:946–952. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources