

Phylodynamic Inference across Epidemic Scales
- PMID: 28204593
- PMCID: PMC5400386
- DOI: 10.1093/molbev/msx077
Phylodynamic Inference across Epidemic Scales
Abstract
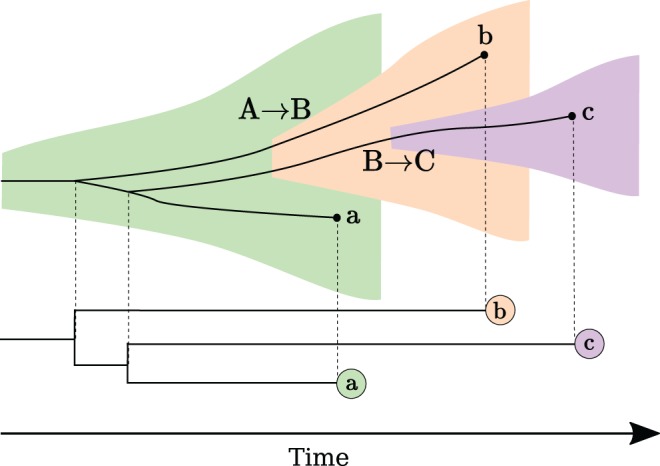
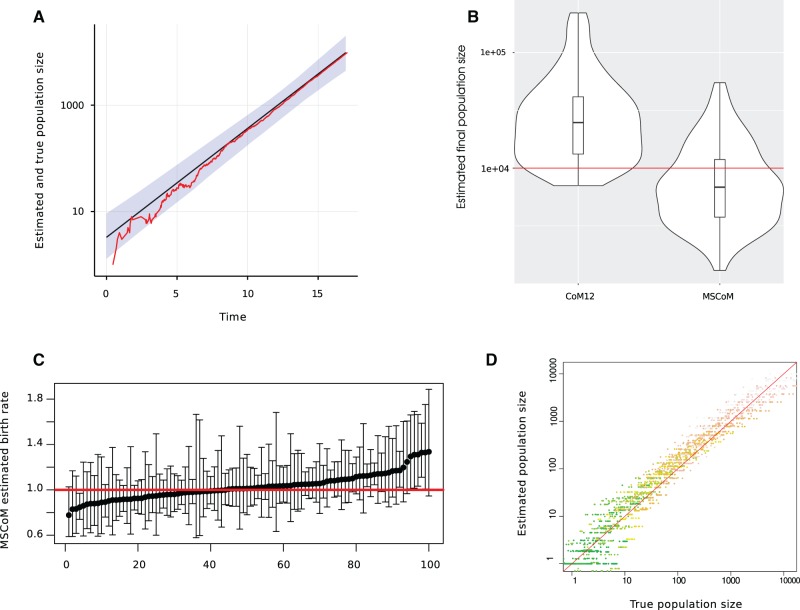
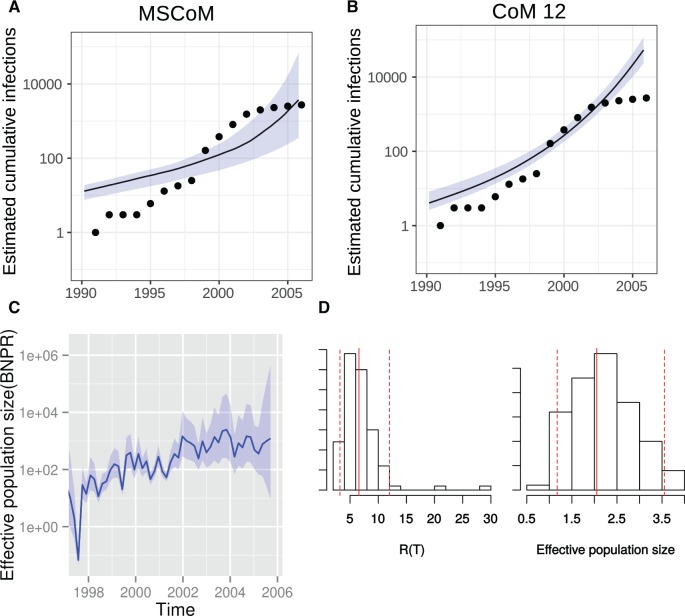
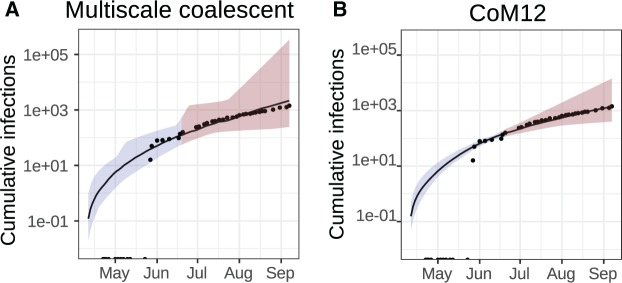
Within-host genetic diversity and large transmission bottlenecks confound phylodynamic inference of epidemiological dynamics. Conventional phylodynamic approaches assume that nodes in a time-scaled pathogen phylogeny correspond closely to the time of transmission between hosts that are ancestral to the sample. However, when hosts harbor diverse pathogen populations, node times can substantially pre-date infection times. Imperfect bottlenecks can cause lineages sampled in different individuals to coalesce in unexpected patterns. To address realistic violations of standard phylodynamic assumptions we developed a new inference approach based on a multi-scale coalescent model, accounting for nonlinear epidemiological dynamics, heterogeneous sampling through time, non-negligible genetic diversity of pathogens within hosts, and imperfect transmission bottlenecks. We apply this method to HIV-1 and Ebola virus (EBOV) outbreak sequence data, illustrating how and when conventional phylodynamic inference may give misleading results. Within-host diversity of HIV-1 causes substantial upwards bias in the number of infected hosts using conventional coalescent models, but estimates using the multi-scale model have greater consistency with reported number of diagnoses through time. In contrast, we find that within-host diversity of EBOV has little influence on estimated numbers of infected hosts or reproduction numbers, and estimates are highly consistent with the reported number of diagnoses through time. The multi-scale coalescent also enables estimation of within-host effective population size using single sequences from a random sample of patients. We find within-host population genetic diversity of HIV-1 p17 to be 2Nμ=0.012 (95% CI 0.0066-0.023), which is lower than estimates based on HIV envelope serial sequencing of individual patients.
Keywords: Ebola; HIV; coalescent; phylodynamics.
© The Author 2017. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
References
-
- Akima H. 1970. A new method of interpolation and smooth curve fitting based on local procedures. J ACM (JACM) 17:589–602.
-
- Anderson RM, May RM, Anderson B.. 1992. Infectious diseases of humans: dynamics and control, volume 28 Wiley Online Library.
-
- Balode D, Ferdats A, Dievberna I, Viksna L, Rozentale B, Kolupajeva T, Konicheva V, Leitner T.. 2004. Rapid epidemic spread of HIV type 1 subtype A1 among intravenous drug users in Latvia and slower spread of subtype B among other risk groups. AIDS Res Hum Retroviruses 20:245–249. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
