

MARV: a tool for genome-wide multi-phenotype analysis of rare variants
- PMID: 28209135
- PMCID: PMC5311849
- DOI: 10.1186/s12859-017-1530-2
MARV: a tool for genome-wide multi-phenotype analysis of rare variants
Abstract
Background: Genome-wide association studies have enabled identification of thousands of loci for hundreds of traits. Yet, for most human traits a substantial part of the estimated heritability is unexplained. This and recent advances in technology to produce high-dimensional data cost-effectively have led to method development beyond standard common variant analysis, including single-phenotype rare variant and multi-phenotype common variant analysis, with the latter increasing power for locus discovery and providing suggestions of pleiotropic effects. However, there are currently no optimal methods and tools for the combined analysis of rare variants and multiple phenotypes.
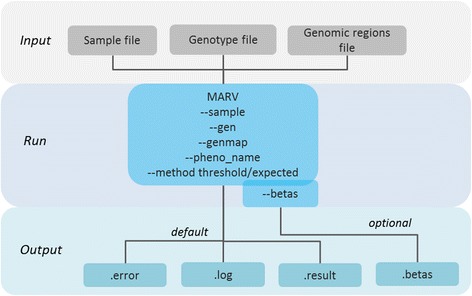
Results: We propose a user-friendly software tool MARV for Multi-phenotype Analysis of Rare Variants. The tool is based on a method that collapses rare variants within a genomic region and models the proportion of minor alleles in the rare variants on a linear combination of multiple phenotypes. MARV provides analyses of all phenotype combinations within one run and calculates the Bayesian Information Criterion to facilitate model selection. The running time increases with the size of the genetic data while the number of phenotypes to analyse has little effect both on running time and required memory. We illustrate the use of MARV with analysis of triglycerides (TG), fasting insulin (FI) and waist-to-hip ratio (WHR) in 4,721 individuals from the Northern Finland Birth Cohort 1966. The analysis suggests novel multi-phenotype effects for these metabolic traits at APOA5 and ZNF259, and at ZNF259 provides stronger support for association (P TG+FI = 1.8 × 10-9) than observed in single phenotype rare variant analyses (P TG = 6.5 × 10-8 and P FI = 0.27).
Conclusions: MARV is a computationally efficient, flexible and user-friendly software tool allowing rapid identification of rare variant effects on multiple phenotypes, thus paving the way for novel discoveries and insights into biology of complex traits.
Keywords: High-dimensional data; Multi-phenotype analysis; Rare variant analysis.
Figures



Similar articles
-
A rare-variant test for high-dimensional data.Eur J Hum Genet. 2017 Aug;25(8):988-994. doi: 10.1038/ejhg.2017.90. Epub 2017 May 24. Eur J Hum Genet. 2017. PMID: 28537275 Free PMC article.
-
A visual and curatorial approach to clinical variant prioritization and disease gene discovery in genome-wide diagnostics.Genome Med. 2016 Feb 2;8(1):13. doi: 10.1186/s13073-016-0261-8. Genome Med. 2016. PMID: 26838676 Free PMC article.
-
Optimized phenotyping of complex morphological traits: enhancing discovery of common and rare genetic variants.Brief Bioinform. 2025 Mar 4;26(2):bbaf090. doi: 10.1093/bib/bbaf090. Brief Bioinform. 2025. PMID: 40062617 Free PMC article.
-
Molecular genetic studies of complex phenotypes.Transl Res. 2012 Feb;159(2):64-79. doi: 10.1016/j.trsl.2011.08.001. Epub 2011 Aug 31. Transl Res. 2012. PMID: 22243791 Free PMC article. Review.
-
Rare-variant genome-wide association studies: a new frontier in genetic analysis of complex traits.Pharmacogenomics. 2013 Mar;14(4):413-24. doi: 10.2217/pgs.13.36. Pharmacogenomics. 2013. PMID: 23438888 Review.
Cited by
-
Multi-Phenotype Association Decomposition: Unraveling Complex Gene-Phenotype Relationships.Front Genet. 2019 May 10;10:417. doi: 10.3389/fgene.2019.00417. eCollection 2019. Front Genet. 2019. PMID: 31134130 Free PMC article.
-
Variant Impact Predictor database (VIPdb), version 2: Trends from 25 years of genetic variant impact predictors.bioRxiv [Preprint]. 2024 Jun 28:2024.06.25.600283. doi: 10.1101/2024.06.25.600283. bioRxiv. 2024. Update in: Hum Genomics. 2024 Aug 28;18(1):90. doi: 10.1186/s40246-024-00663-z. PMID: 38979289 Free PMC article. Updated. Preprint.
-
Bivariate quantitative Bayesian LASSO for detecting association of rare haplotypes with two correlated continuous phenotypes.Front Genet. 2023 Mar 9;14:1104727. doi: 10.3389/fgene.2023.1104727. eCollection 2023. Front Genet. 2023. PMID: 36968609 Free PMC article.
-
Breeding for Climate Change Resilience: A Case Study of Loblolly Pine (Pinus taeda L.) in North America.Front Plant Sci. 2021 Apr 30;12:606908. doi: 10.3389/fpls.2021.606908. eCollection 2021. Front Plant Sci. 2021. PMID: 33995428 Free PMC article. Review.
-
Pathway analysis for genome-wide genetic variation data: Analytic principles, latest developments, and new opportunities.J Genet Genomics. 2021 Mar 20;48(3):173-183. doi: 10.1016/j.jgg.2021.01.007. Epub 2021 Feb 26. J Genet Genomics. 2021. PMID: 33896739 Free PMC article. Review.
References
-
- McVean GA, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, Flicek P, Gabriel SB, Gibbs RA, Green ED, Hurles ME, Knoppers BM, Korbel JO, Lander ES, Lee C, Lehrach H, Mardis ER, Marth GT, McVean GA, Nickerson DA, Schmidt JP, Sherry ST, Wang J, Wilson RK, Gibbs RA, Dinh H, Kovar C, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(V):56–65. doi: 10.1038/nature11632. - DOI - PMC - PubMed
-
- Walter K, Min JL, Huang J, Crooks L, Memari Y, McCarthy S, Perry JRB, Xu C, Futema M, Lawson D, Iotchkova V, Schiffels S, Hendricks AE, Danecek P, Li R, Floyd J, Wain LV, Barroso I, Humphries SE, Hurles ME, Zeggini E, Barrett JC, Plagnol V, Brent Richards J, Greenwood CMT, Timpson NJ, Durbin R, Soranzo N, Bala S, Clapham P, et al. The UK10K project identifies rare variants in health and disease. Nature. 2015;526:82–90. doi: 10.1038/nature14962. - DOI - PMC - PubMed
-
- The Haplotype Reference Consortium [http://www.haplotype-reference-consortium.org/home]. Accessed 8 Feb 2017.
-
- Huang J, Howie B, McCarthy S, Memari Y, Walter K, Min JL, Danecek P, Malerba G, Trabetti E, Zheng H-F, UK10K Consortium. Gambaro G, Richards JB, Durbin R, Timpson NJ, Marchini J, Soranzo N. Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nat Commun. 2015;6:8111. doi: 10.1038/ncomms9111. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
