

Pathogenic mutations in retinitis pigmentosa 2 predominantly result in loss of RP2 protein stability in humans and zebrafish
- PMID: 28209709
- PMCID: PMC5391753
- DOI: 10.1074/jbc.M116.760314
Pathogenic mutations in retinitis pigmentosa 2 predominantly result in loss of RP2 protein stability in humans and zebrafish
Abstract
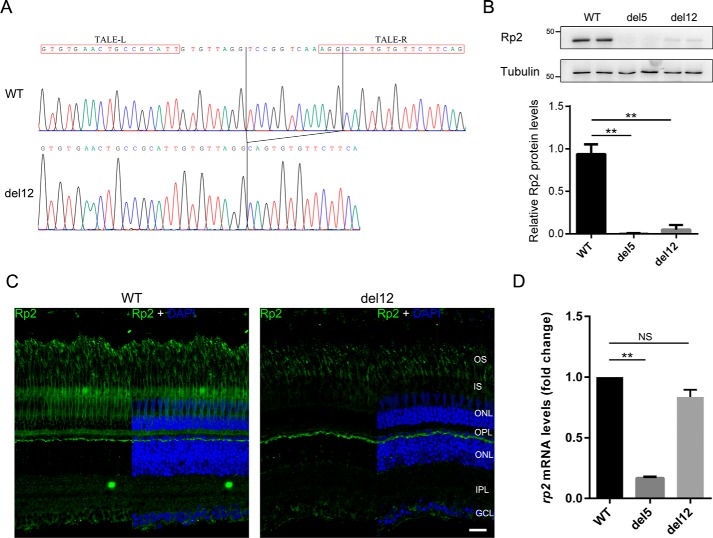
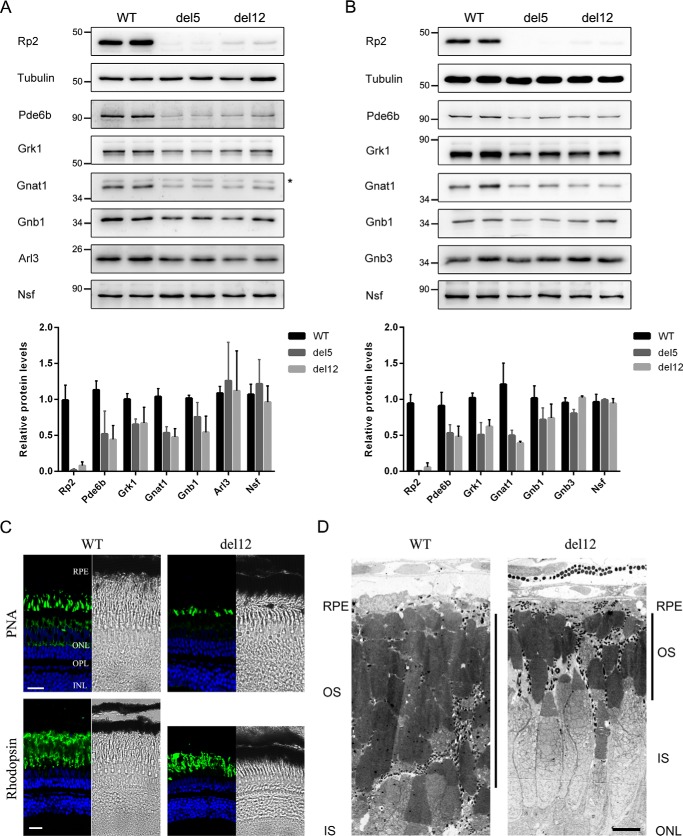
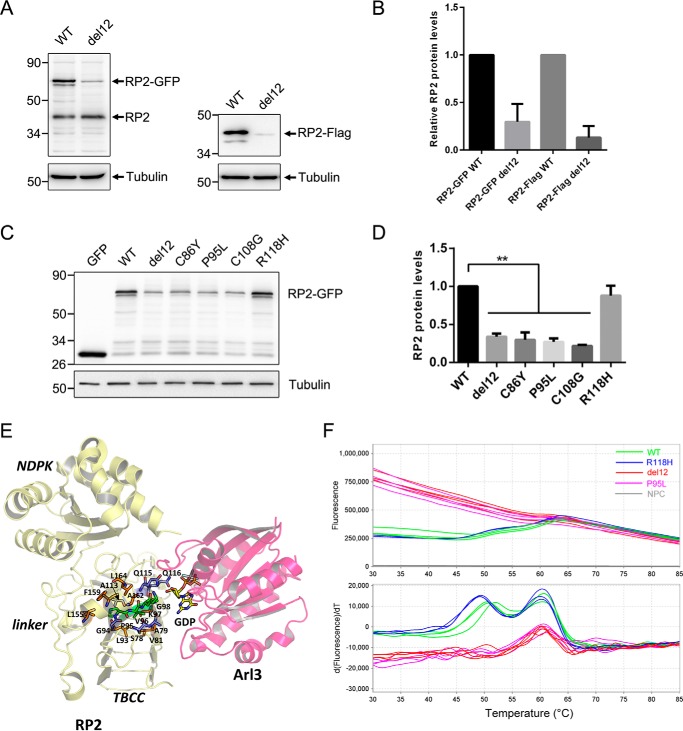
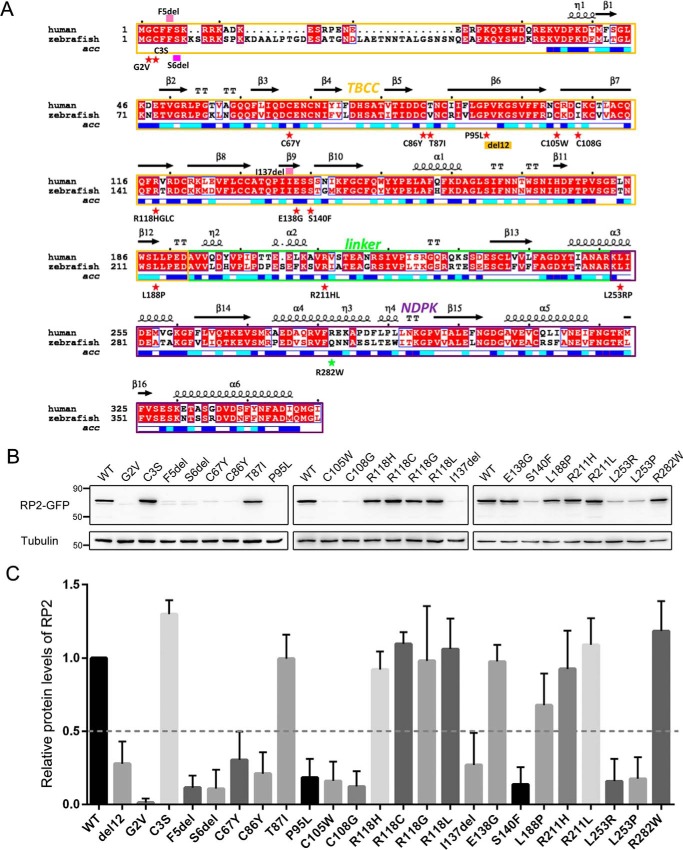
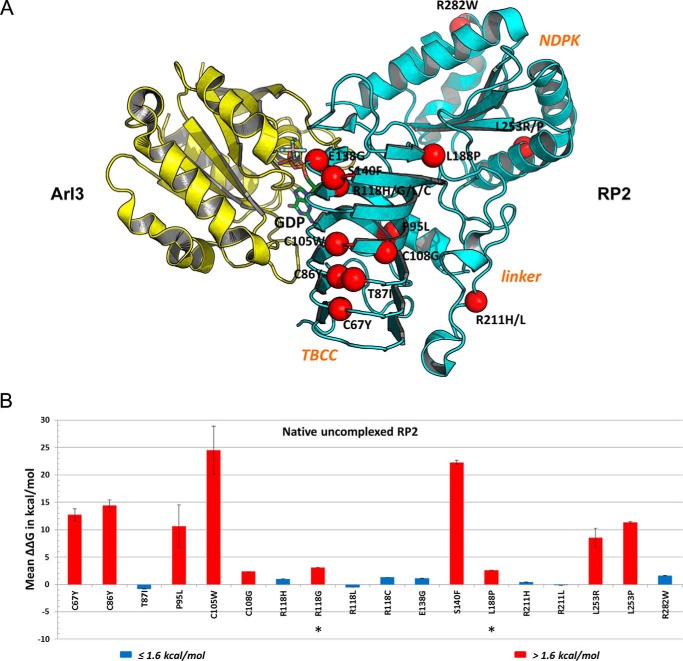
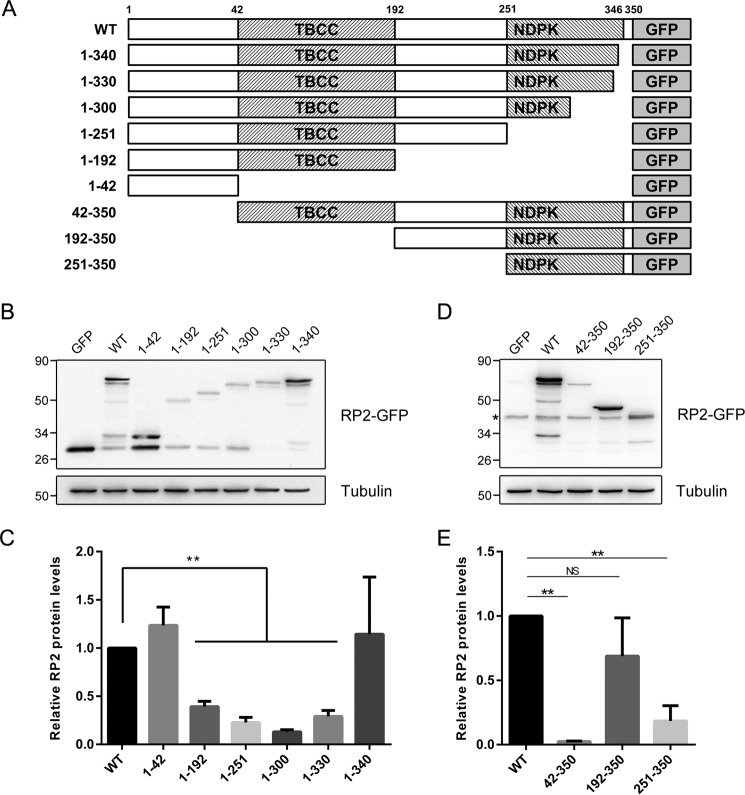
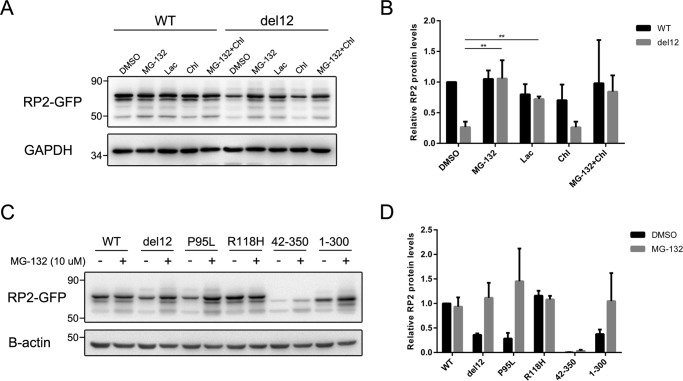
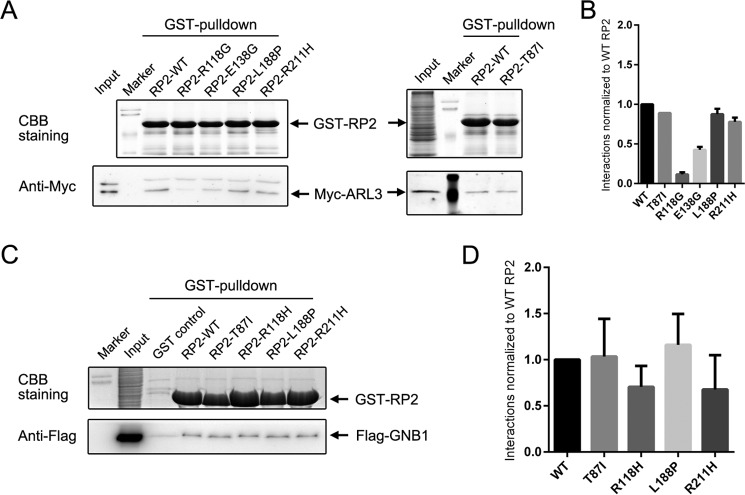
Mutations in retinitis pigmentosa 2 (RP2) account for 10-20% of X-linked retinitis pigmentosa (RP) cases. The encoded RP2 protein is implicated in ciliary trafficking of myristoylated and prenylated proteins in photoreceptor cells. To date >70 mutations in RP2 have been identified. How these mutations disrupt the function of RP2 is not fully understood. Here we report a novel in-frame 12-bp deletion (c.357_368del, p.Pro120_Gly123del) in zebrafish rp2 The mutant zebrafish shows reduced rod phototransduction proteins and progressive retinal degeneration. Interestingly, the protein level of mutant Rp2 is almost undetectable, whereas its mRNA level is near normal, indicating a possible post-translational effect of the mutation. Consistent with this hypothesis, the equivalent 12-bp deletion in human RP2 markedly impairs RP2 protein stability and reduces its protein level. Furthermore, we found that a majority of the RP2 pathogenic mutations (including missense, single-residue deletion, and C-terminal truncation mutations) severely destabilize the RP2 protein. The destabilized RP2 mutant proteins are degraded via the proteasome pathway, resulting in dramatically decreased protein levels. The remaining non-destabilizing mutations T87I, R118H/R118G/R118L/R118C, E138G, and R211H/R211L are suggested to impair the interaction between RP2 and its protein partners (such as ARL3) or with as yet unknown partners. By utilizing a combination of in silico, in vitro, and in vivo approaches, our work comprehensively indicates that loss of RP2 protein structural stability is the predominating pathogenic consequence for most RP2 mutations. Our study also reveals a role of the C-terminal domain of RP2 in maintaining the overall protein stability.
Keywords: RP2; mutant; protein degradation; protein stability; retinal degeneration; structural model; transcription activator-like effector nuclease (TALEN); zebrafish.
© 2017 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Hartong D. T., Berson E. L., and Dryja T. P. (2006) Retinitis pigmentosa. Lancet 368, 1795–1809 - PubMed
-
- Prokisch H., Hartig M., Hellinger R., Meitinger T., and Rosenberg T. (2007) A population-based epidemiological and genetic study of X-linked retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 48, 4012–4018 - PubMed
-
- Pelletier V., Jambou M., Delphin N., Zinovieva E., Stum M., Gigarel N., Dollfus H., Hamel C., Toutain A., Dufier J. L., Roche O., Munnich A., Bonnefont J. P., Kaplan J., and Rozet J. M. (2007) Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: genotype-phenotype correlations and impact on genetic counseling. Hum. Mutat. 28, 81–91 - PubMed
-
- Li L., Khan N., Hurd T., Ghosh A. K., Cheng C., Molday R., Heckenlively J. R., Swaroop A., and Khanna H. (2013) Ablation of the X-linked retinitis pigmentosa 2 (Rp2) gene in mice results in opsin mislocalization and photoreceptor degeneration. Invest. Ophthalmol. Vis. Sci. 54, 4503–4511 - PMC - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
