

Lipid transport by TMEM24 at ER-plasma membrane contacts regulates pulsatile insulin secretion
- PMID: 28209843
- PMCID: PMC5414417
- DOI: 10.1126/science.aah6171
Lipid transport by TMEM24 at ER-plasma membrane contacts regulates pulsatile insulin secretion
Abstract
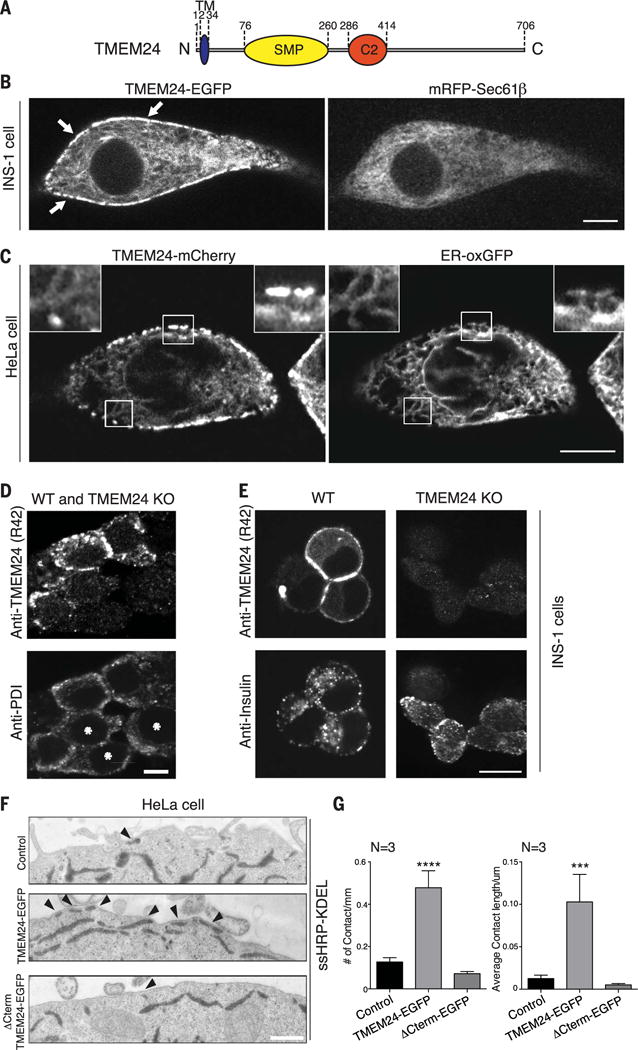
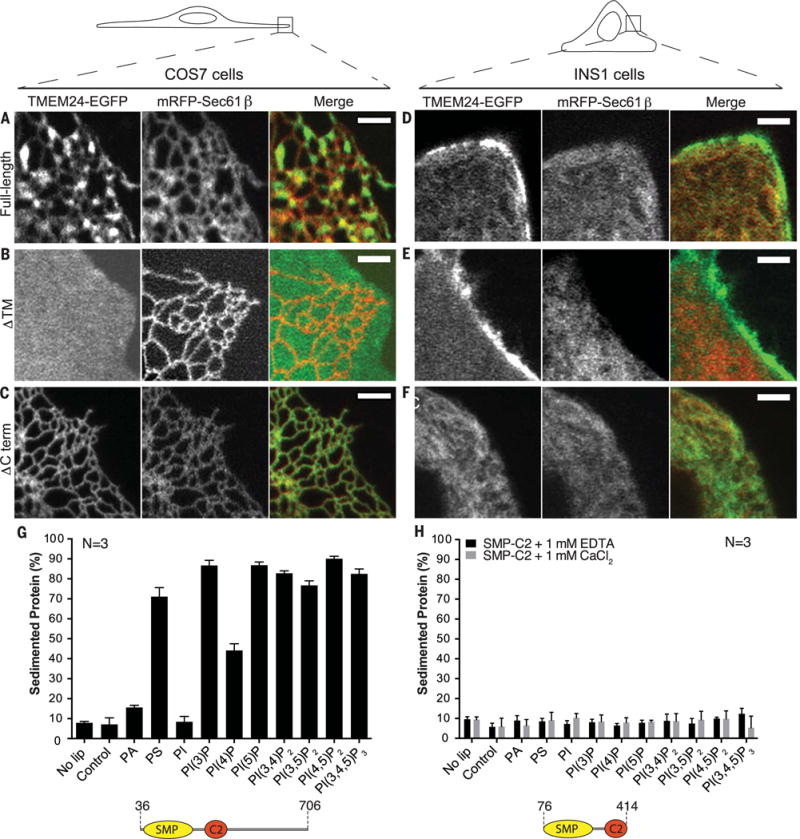
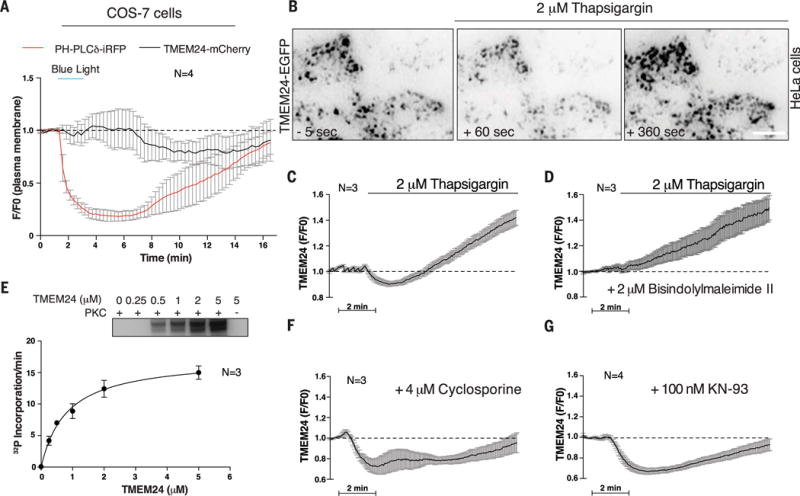
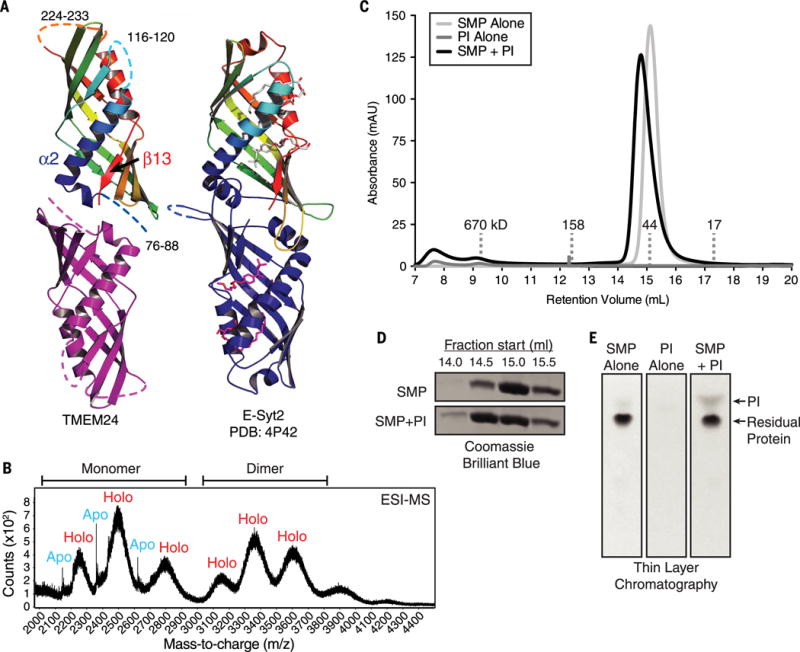
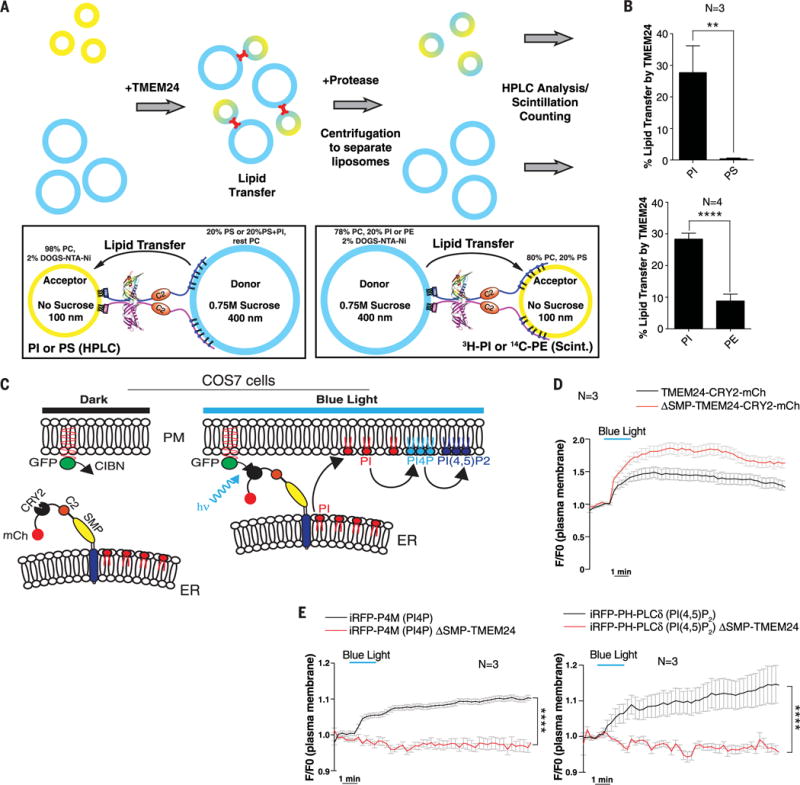
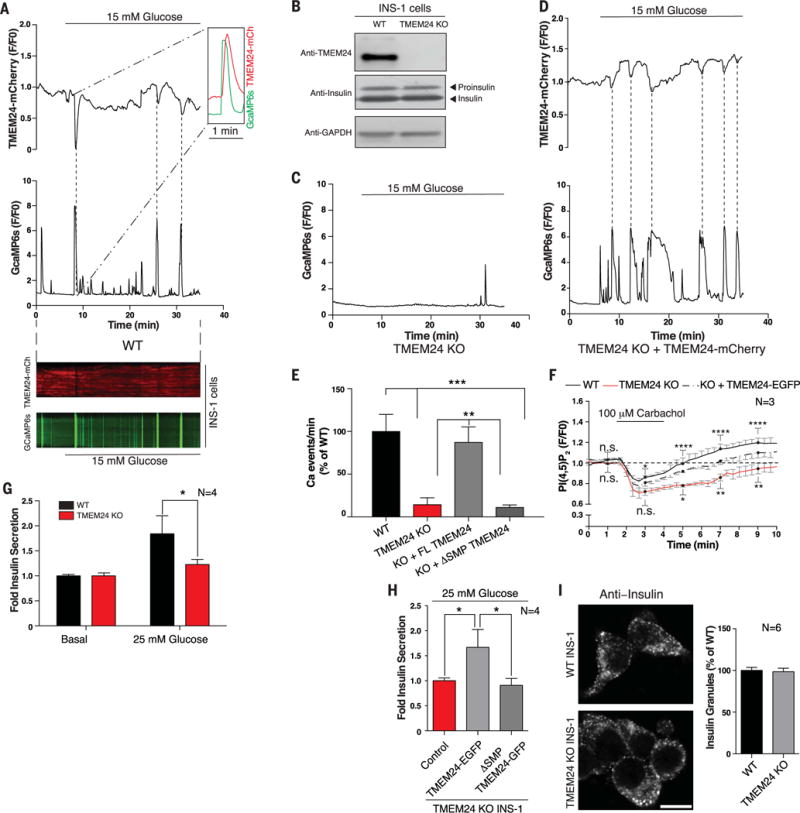
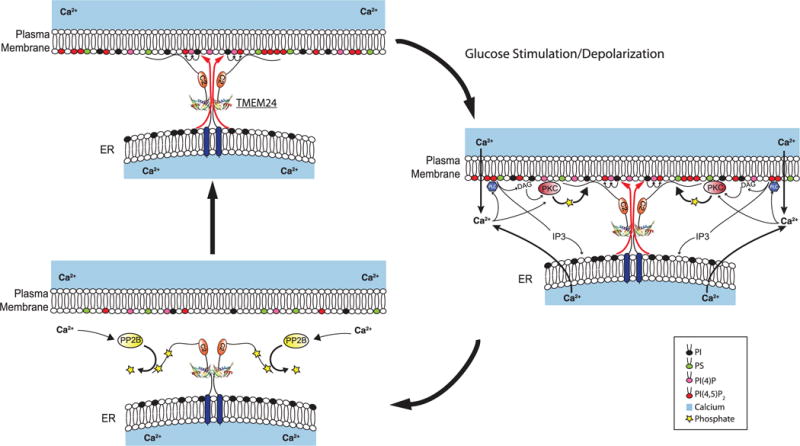
Insulin is released by β cells in pulses regulated by calcium and phosphoinositide signaling. Here, we describe how transmembrane protein 24 (TMEM24) helps coordinate these signaling events. We showed that TMEM24 is an endoplasmic reticulum (ER)-anchored membrane protein whose reversible localization to ER-plasma membrane (PM) contacts is governed by phosphorylation and dephosphorylation in response to oscillations in cytosolic calcium. A lipid-binding module in TMEM24 transports the phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] precursor phosphatidylinositol between bilayers, allowing replenishment of PI(4,5)P2 hydrolyzed during signaling. In the absence of TMEM24, calcium oscillations are abolished, leading to a defect in triggered insulin release. Our findings implicate direct lipid transport between the ER and the PM in the control of insulin secretion, a process impaired in patients with type II diabetes.
Copyright © 2017, American Association for the Advancement of Science.
Figures
Comment in
-
Papers of note in Science355 (6326).Sci Signal. 2017 Feb 21;10(467):eaam9937. doi: 10.1126/scisignal.aam9937. Sci Signal. 2017. PMID: 28223417
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
