

APP as a Protective Factor in Acute Neuronal Insults
- PMID: 28210211
- PMCID: PMC5288400
- DOI: 10.3389/fnmol.2017.00022
APP as a Protective Factor in Acute Neuronal Insults
Abstract
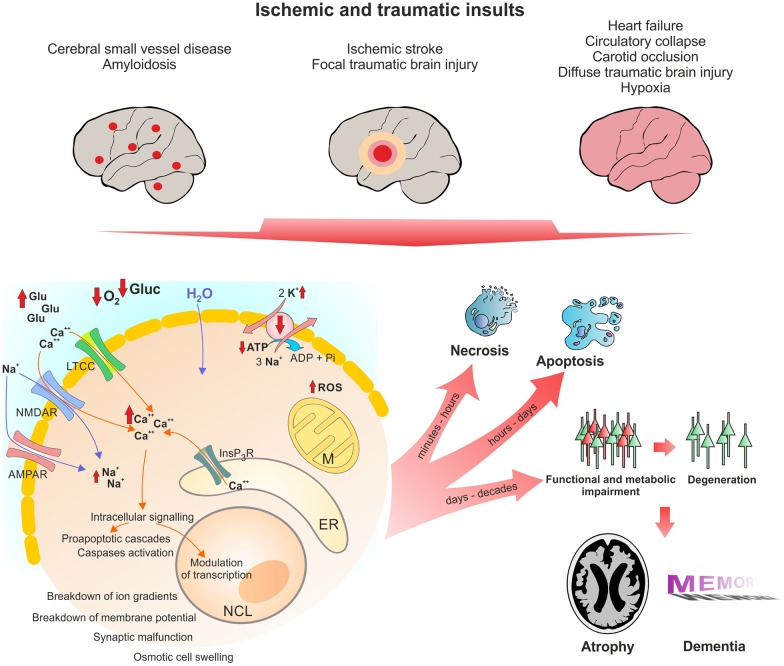
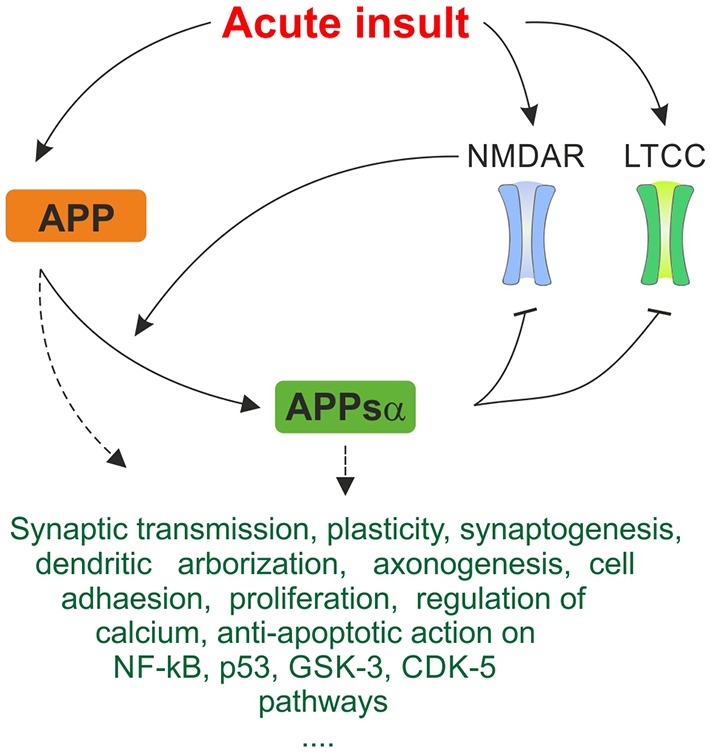
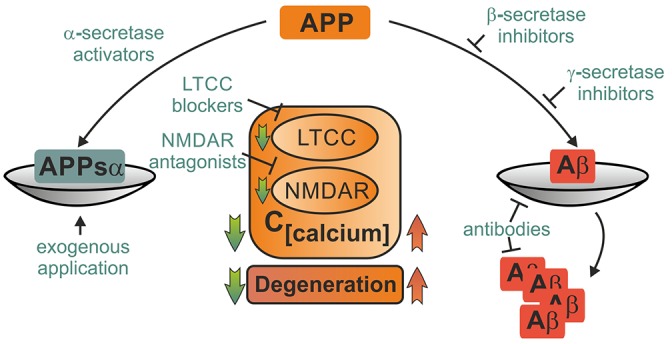
Despite its key role in the molecular pathology of Alzheimer's disease (AD), the physiological function of amyloid precursor protein (APP) is unknown. Increasing evidence, however, points towards a neuroprotective role of this membrane protein in situations of metabolic stress. A key observation is the up-regulation of APP following acute (stroke, cardiac arrest) or chronic (cerebrovascular disease) hypoxic-ischemic conditions. While this mechanism may increase the risk or severity of AD, APP by itself or its soluble extracellular fragment APPsα can promote neuronal survival. Indeed, different animal models of acute hypoxia-ischemia, traumatic brain injury (TBI) and excitotoxicity have revealed protective effects of APP or APPsα. The underlying mechanisms involve APP-mediated regulation of calcium homeostasis via NMDA receptors (NMDAR), voltage-gated calcium channels (VGCC) or internal calcium stores. In addition, APP affects the expression of survival- or apoptosis-related genes as well as neurotrophic factors. In this review, we summarize the current understanding of the neuroprotective role of APP and APPsα and possible implications for future research and new therapeutic strategies.
Keywords: Alzheimer; amyloid precursor protein; calcium toxicity; cell death; ischemia; neuroprotection; stroke; traumatic brain injury.
Figures
References
-
- Alzheimer A. (1906). Über einen eigenartigen, schweren Erkrankungsprozeß der Hirnrinde. Neurol. Cent. 23, 1129–1136.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources