

CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression
- PMID: 28215704
- PMCID: PMC5328582
- DOI: 10.1016/j.cell.2017.01.021
CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression
Abstract
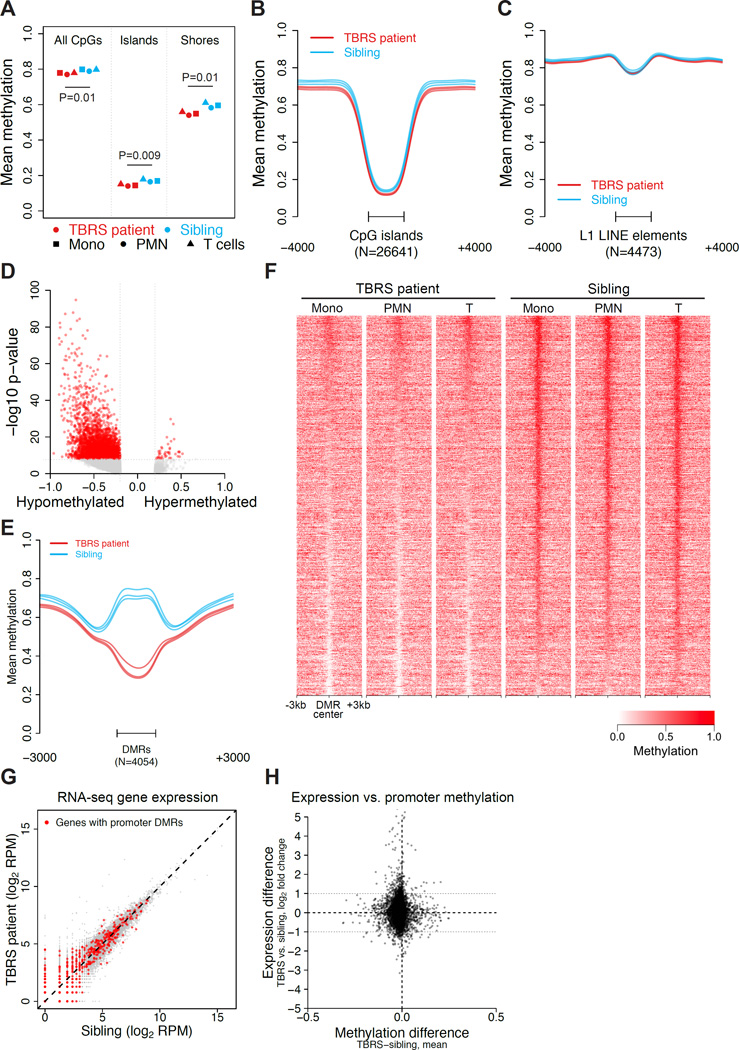
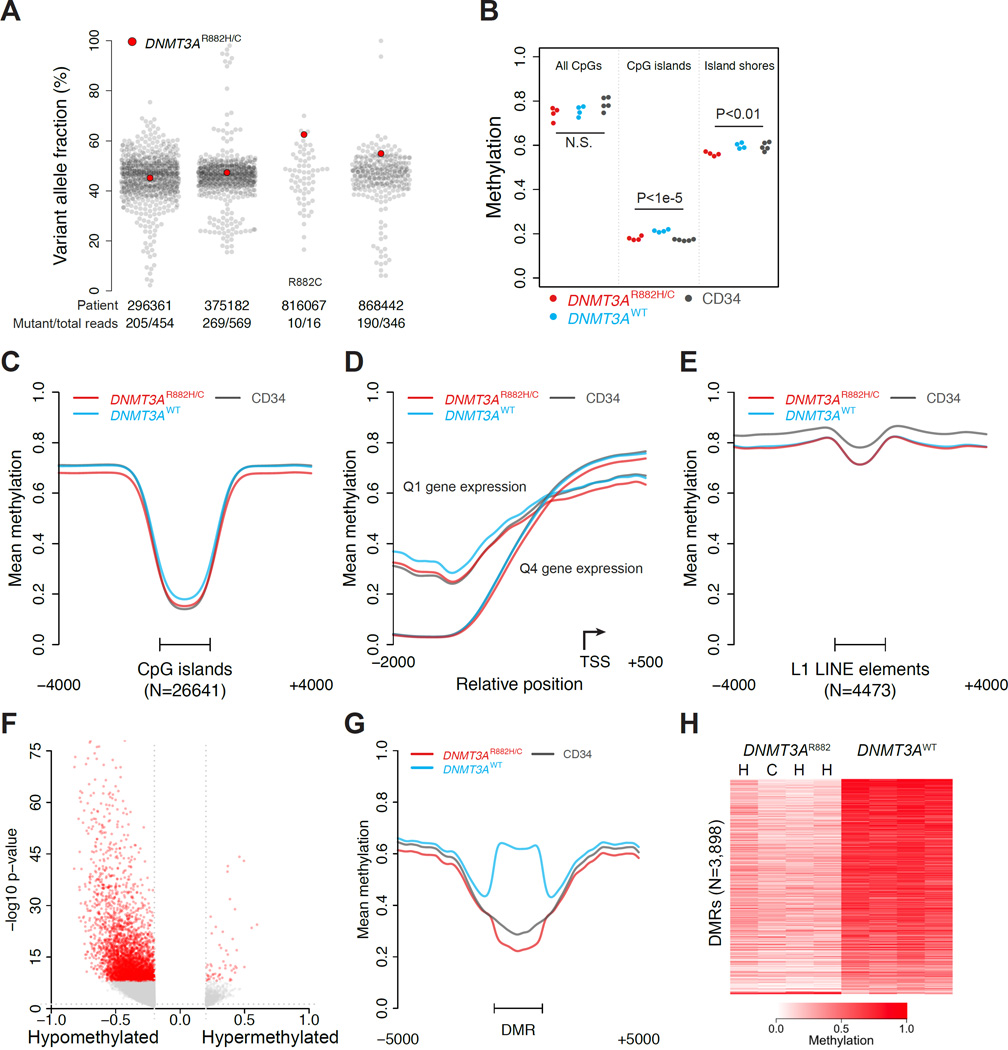
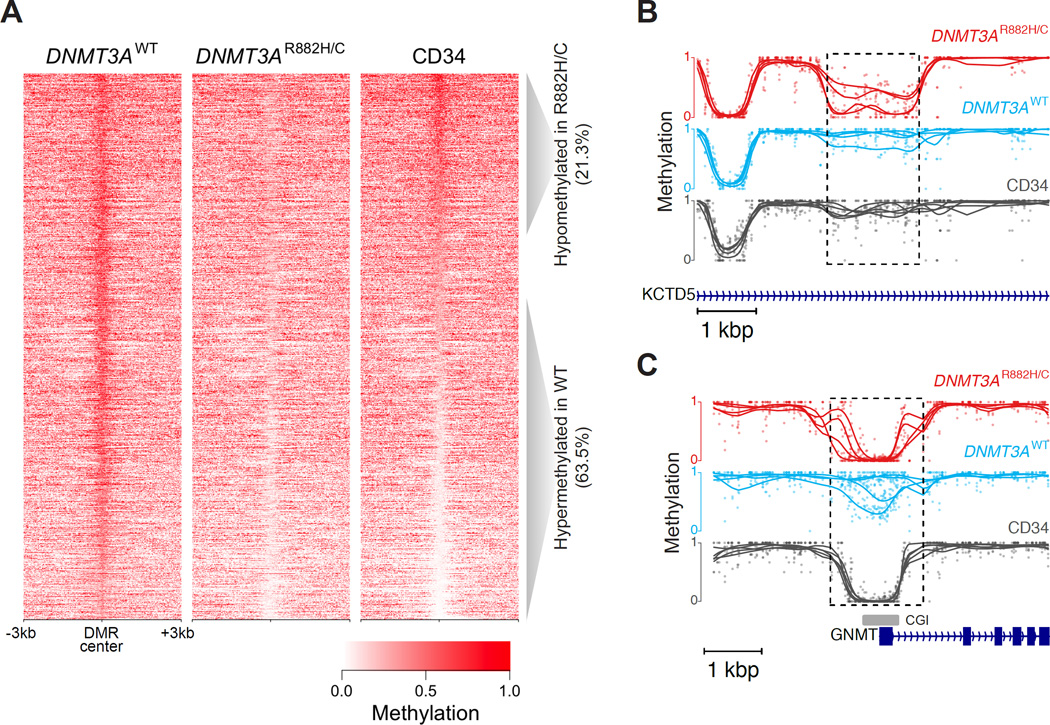
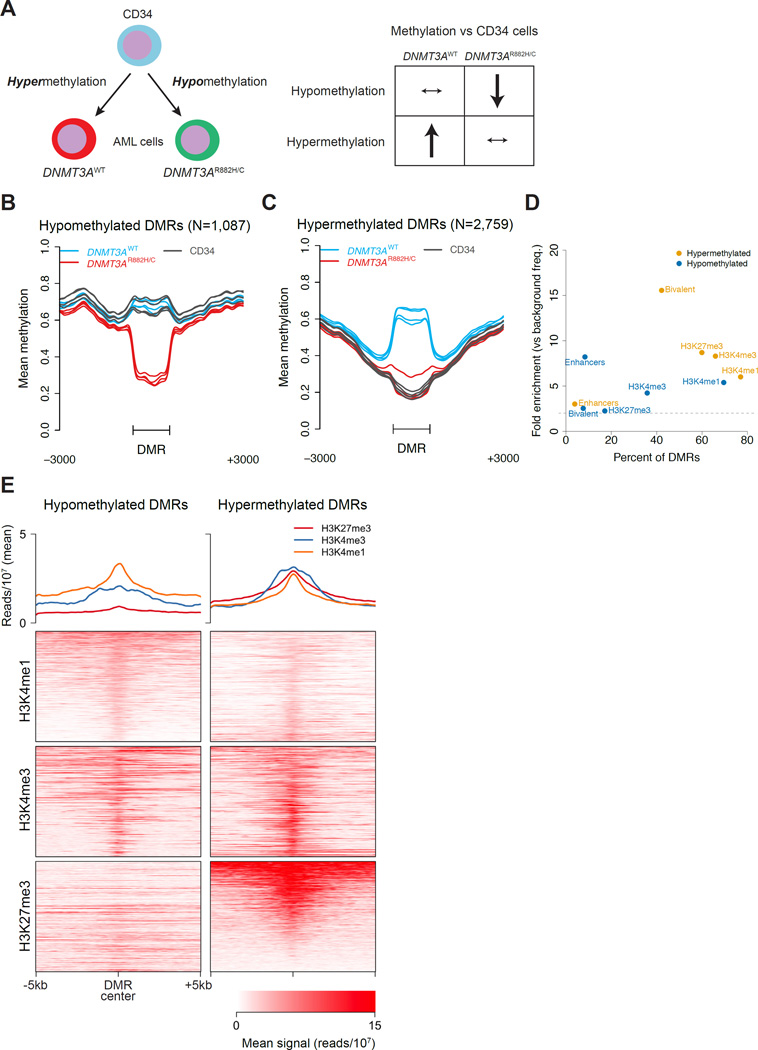
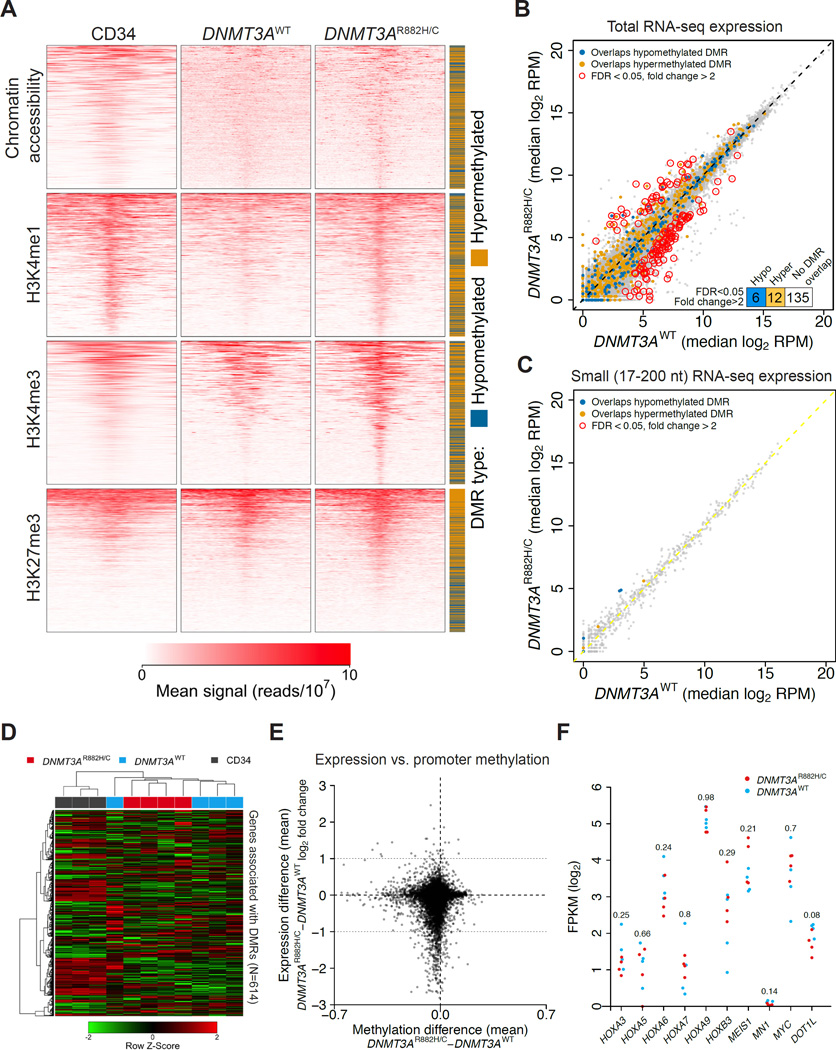
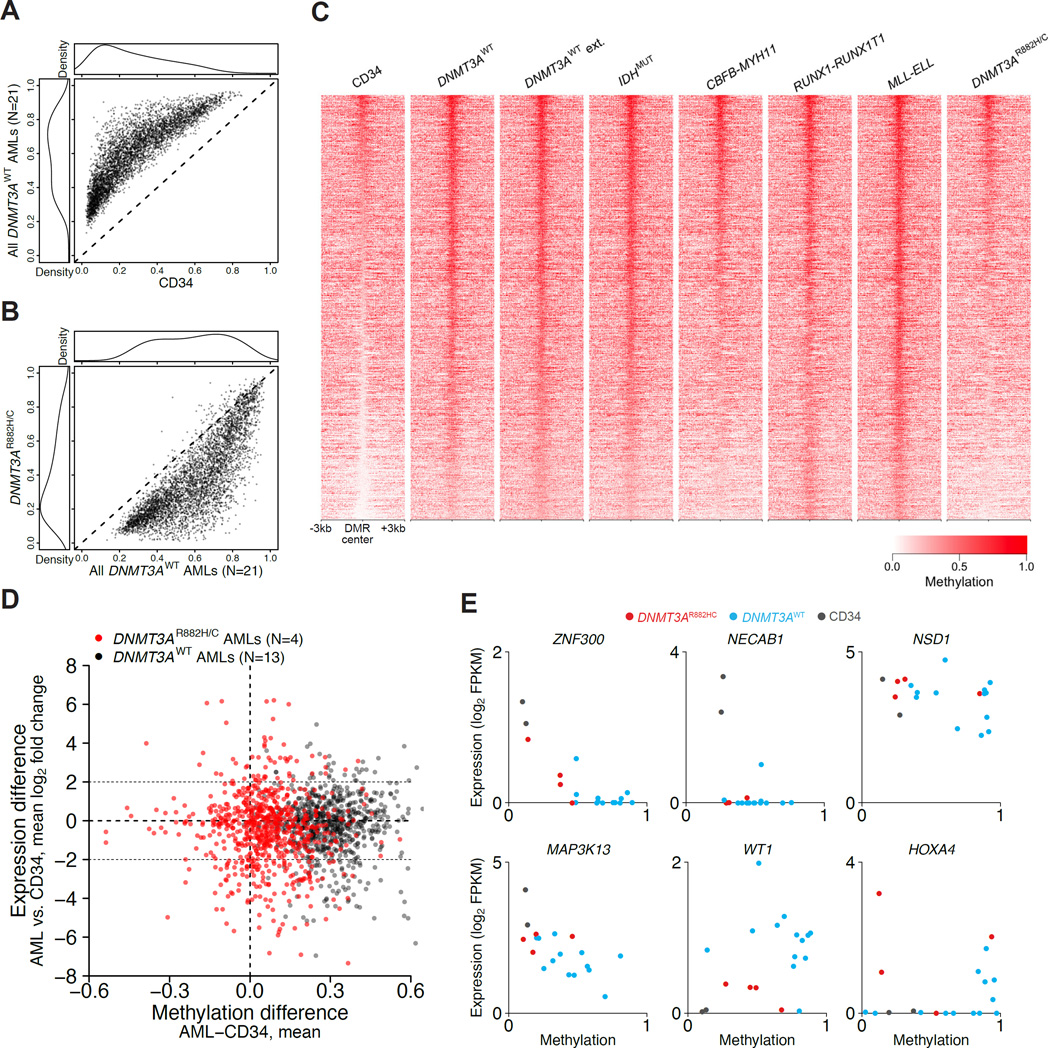
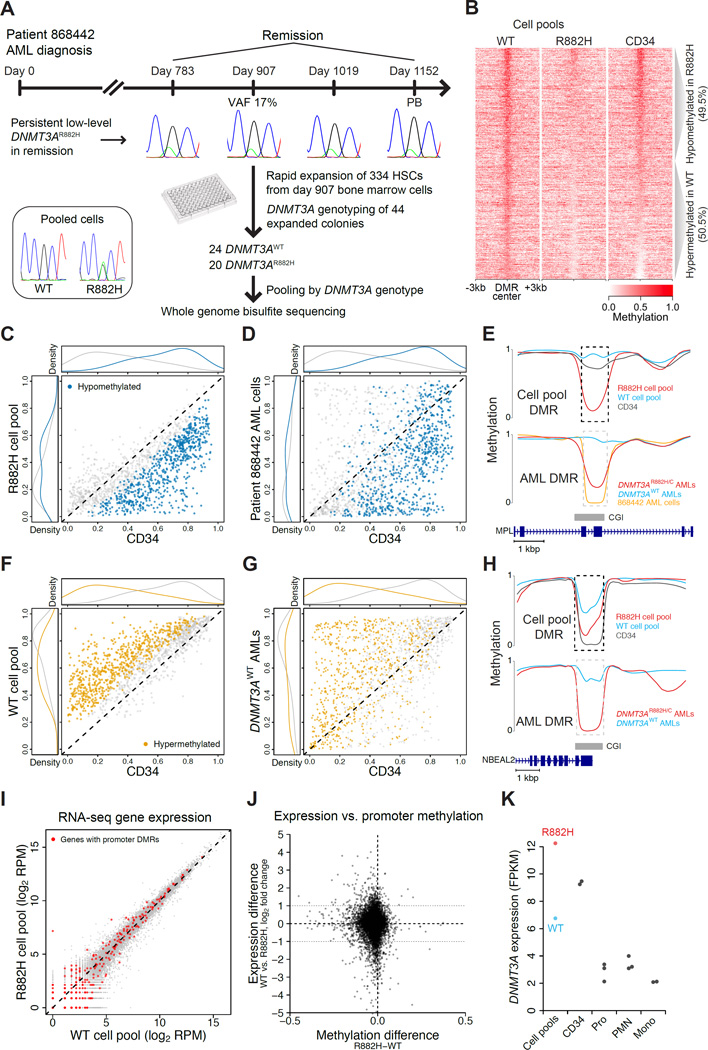
DNMT3A mutations occur in ∼25% of acute myeloid leukemia (AML) patients. The most common mutation, DNMT3AR882H, has dominant negative activity that reduces DNA methylation activity by ∼80% in vitro. To understand the contribution of DNMT3A-dependent methylation to leukemogenesis, we performed whole-genome bisulfite sequencing of primary leukemic and non-leukemic cells in patients with or without DNMT3AR882 mutations. Non-leukemic hematopoietic cells with DNMT3AR882H displayed focal methylation loss, suggesting that hypomethylation antedates AML. Although virtually all AMLs with wild-type DNMT3A displayed CpG island hypermethylation, this change was not associated with gene silencing and was essentially absent in AMLs with DNMT3AR882 mutations. Primary hematopoietic stem cells expanded with cytokines were hypermethylated in a DNMT3A-dependent manner, suggesting that hypermethylation may be a response to, rather than a cause of, cellular proliferation. Our findings suggest that hypomethylation is an initiating phenotype in AMLs with DNMT3AR882, while DNMT3A-dependent CpG island hypermethylation is a consequence of AML progression.
Keywords: AML; CpG island hypermethylation; DNA methylation; DNMT3A; leukemia.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Baylin SB, Höppener JW, de Bustros A, Steenbergh PH, Lips CJ, Nelkin BD. DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas. Cancer Research. 1986;46:2917–2922. - PubMed
-
- Baylin S, Bestor TH. Altered methylation patterns in cancer cell genomes: cause or consequence? Cancer Cell. 2002;1:299–305. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
