

KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer
- PMID: 28215705
- PMCID: PMC5541948
- DOI: 10.1016/j.cell.2017.01.020
KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer
Abstract
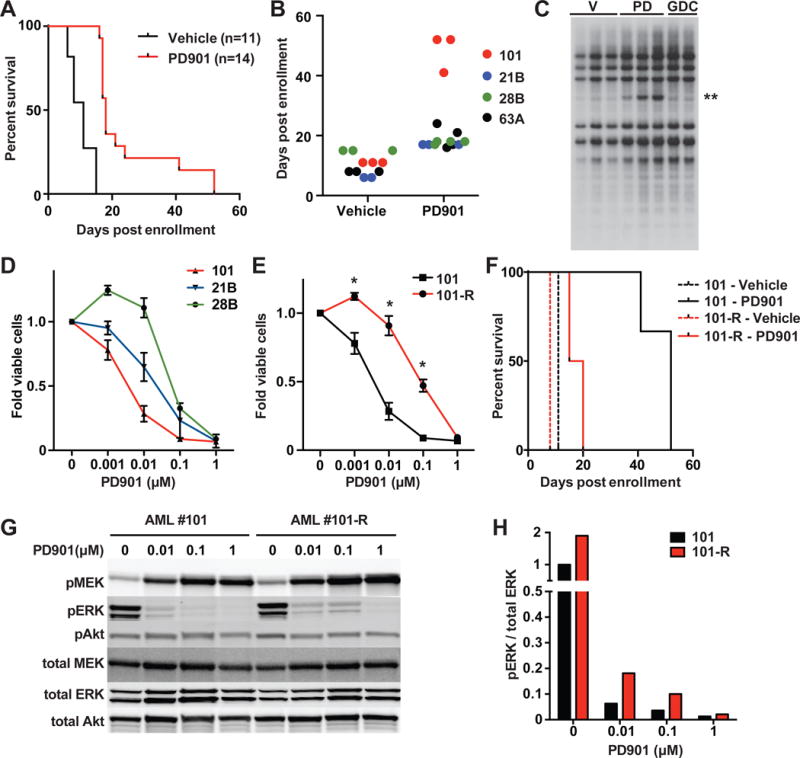
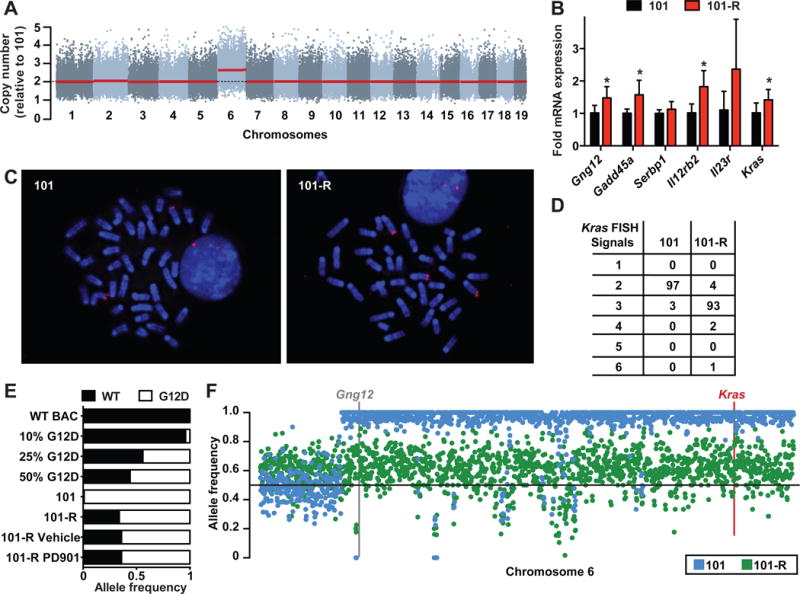
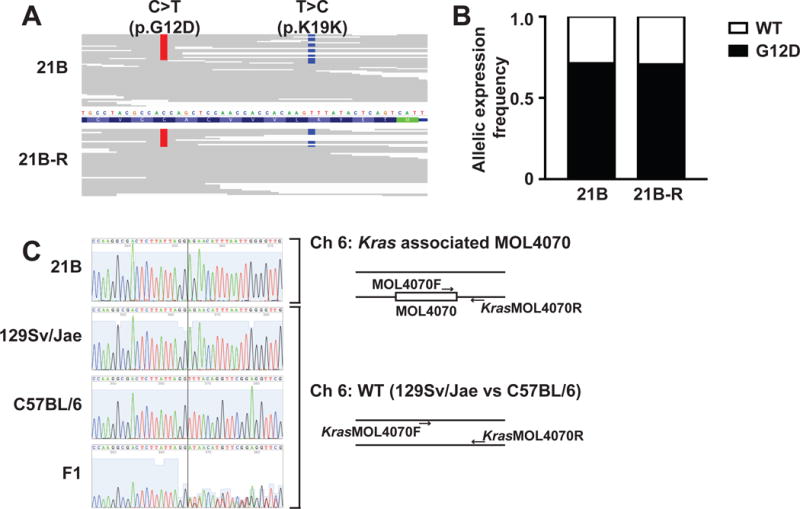
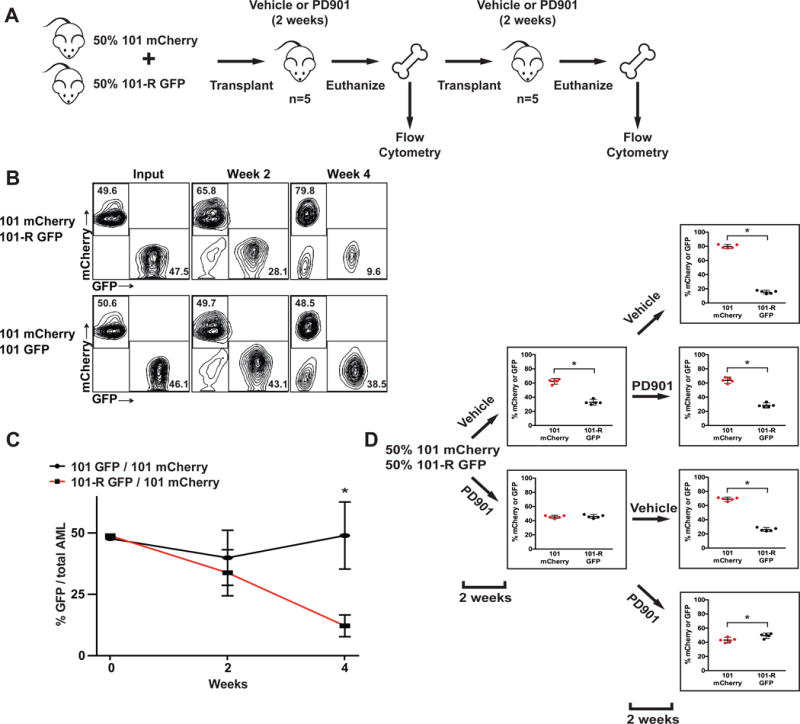
Investigating therapeutic "outliers" that show exceptional responses to anti-cancer treatment can uncover biomarkers of drug sensitivity. We performed preclinical trials investigating primary murine acute myeloid leukemias (AMLs) generated by retroviral insertional mutagenesis in KrasG12D "knockin" mice with the MEK inhibitor PD0325901 (PD901). One outlier AML responded and exhibited intrinsic drug resistance at relapse. Loss of wild-type (WT) Kras enhanced the fitness of the dominant clone and rendered it sensitive to MEK inhibition. Similarly, human colorectal cancer cell lines with increased KRAS mutant allele frequency were more sensitive to MAP kinase inhibition, and CRISPR-Cas9-mediated replacement of WT KRAS with a mutant allele sensitized heterozygous mutant HCT116 cells to treatment. In a prospectively characterized cohort of patients with advanced cancer, 642 of 1,168 (55%) with KRAS mutations exhibited allelic imbalance. These studies demonstrate that serial genetic changes at the Kras/KRAS locus are frequent in cancer and modulate competitive fitness and MEK dependency.
Keywords: AML; KRAS; MEK inhibition; allelic imbalance; colorectal cancer; drug resistance.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Genetics: Fitness depends on KRAS imbalance.Nat Rev Clin Oncol. 2017 May;14(5):262-263. doi: 10.1038/nrclinonc.2017.32. Epub 2017 Mar 7. Nat Rev Clin Oncol. 2017. PMID: 28266521 No abstract available.
-
KRAS Allelic Imbalance: Strengths and Weaknesses in Numbers.Trends Mol Med. 2017 May;23(5):377-378. doi: 10.1016/j.molmed.2017.03.005. Epub 2017 Mar 31. Trends Mol Med. 2017. PMID: 28372922
Comment on
-
Doubling Down on Mutant RAS Can MEK or Break Leukemia.Cell. 2017 Feb 23;168(5):749-750. doi: 10.1016/j.cell.2017.02.013. Cell. 2017. PMID: 28235190
References
-
- Bengtsson H, Simpson K, Bullard J. Tech rep D.o Statistics. University of California; Berkeley: 2008. aroma.affymetrix: A generic framework in R for analyzing small to very large Affymetrix data sets in bounded memory.
-
- Bremner R, Balmain A. Genetic changes in skin tumor progression: correlation between presence of a mutant ras gene and loss of heterozygosity on mouse chromosome 7. Cell. 1990;61:407–417. - PubMed
-
- Brown AP, Carlson TC, Loi CM, Graziano MJ. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemother Pharmacol. 2007;59:671–679. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
