

A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments
- PMID: 28218669
- PMCID: PMC5343975
- DOI: 10.3390/ijms18020441
A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments
Abstract
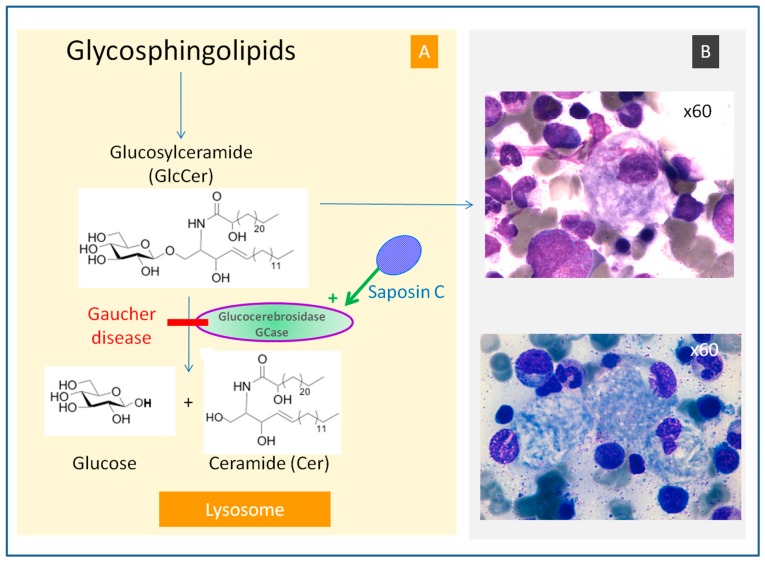
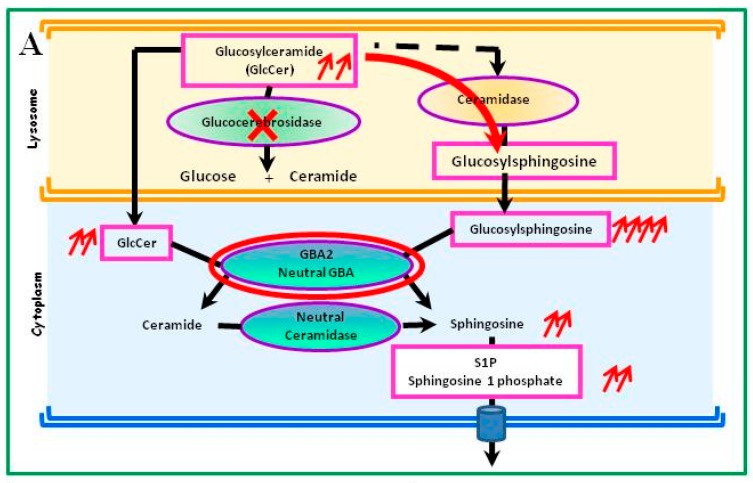
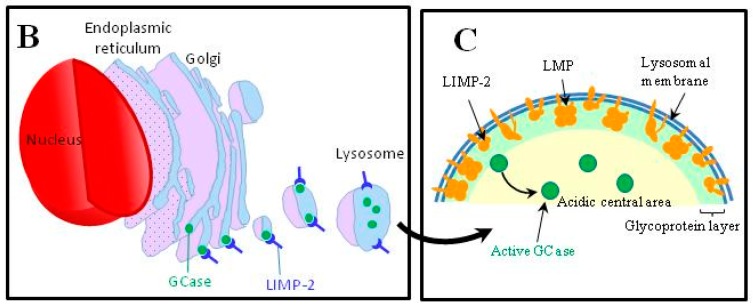
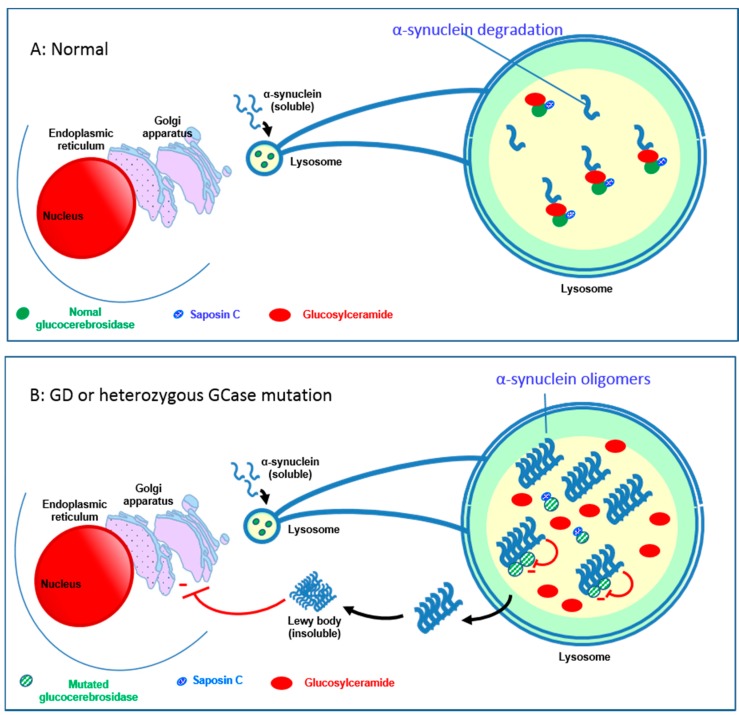
Gaucher disease (GD, ORPHA355) is a rare, autosomal recessive genetic disorder. It is caused by a deficiency of the lysosomal enzyme, glucocerebrosidase, which leads to an accumulation of its substrate, glucosylceramide, in macrophages. In the general population, its incidence is approximately 1/40,000 to 1/60,000 births, rising to 1/800 in Ashkenazi Jews. The main cause of the cytopenia, splenomegaly, hepatomegaly, and bone lesions associated with the disease is considered to be the infiltration of the bone marrow, spleen, and liver by Gaucher cells. Type-1 Gaucher disease, which affects the majority of patients (90% in Europe and USA, but less in other regions), is characterized by effects on the viscera, whereas types 2 and 3 are also associated with neurological impairment, either severe in type 2 or variable in type 3. A diagnosis of GD can be confirmed by demonstrating the deficiency of acid glucocerebrosidase activity in leukocytes. Mutations in the GBA1 gene should be identified as they may be of prognostic value in some cases. Patients with type-1 GD-but also carriers of GBA1 mutation-have been found to be predisposed to developing Parkinson's disease, and the risk of neoplasia associated with the disease is still subject to discussion. Disease-specific treatment consists of intravenous enzyme replacement therapy (ERT) using one of the currently available molecules (imiglucerase, velaglucerase, or taliglucerase). Orally administered inhibitors of glucosylceramide biosynthesis can also be used (miglustat or eliglustat).
Keywords: GBA1 gene; Gaucher disease; biomarkers; enzyme replacement therapy; glucocerebrosidase; lysosomal storage disease; substrate reduction therapy.
Conflict of interest statement
Jérôme Stirnemann has received travel fees from Sanofi-Genzyme. Nadia Belmatoug and Marc G. Berger have received consulting and speaking fees from Sanofi-Genzyme, Shire, Actelion and Pfizer. Roseline Froissart has received travel fees from Sanofi-Genzyme. Grants from these companies were donated to the Department of Clinical Research of the Assistance-Publique Hôpitaux de Paris and of the University Hospital of Clermont-Ferrand. Christine Serratrice and Fabrice Camou have received consultation and speaking fees from Sanofi-Genzyme and Shire. Thierry Levade received support for travel fees from Sanofi-Genzyme and Shire.
Figures
References
-
- Vaccaro A.M., Motta M., Tatti M., Scarpa S., Masuelli L., Bhat M., Vanier M.T., Tylki-Szymanska A., Salvioli R. Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum. Mol. Genet. 2010;19:2987–2997. doi: 10.1093/hmg/ddq204. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
