

An integrative approach unveils FOSL1 as an oncogene vulnerability in KRAS-driven lung and pancreatic cancer
- PMID: 28220783
- PMCID: PMC5321758
- DOI: 10.1038/ncomms14294
An integrative approach unveils FOSL1 as an oncogene vulnerability in KRAS-driven lung and pancreatic cancer
Abstract
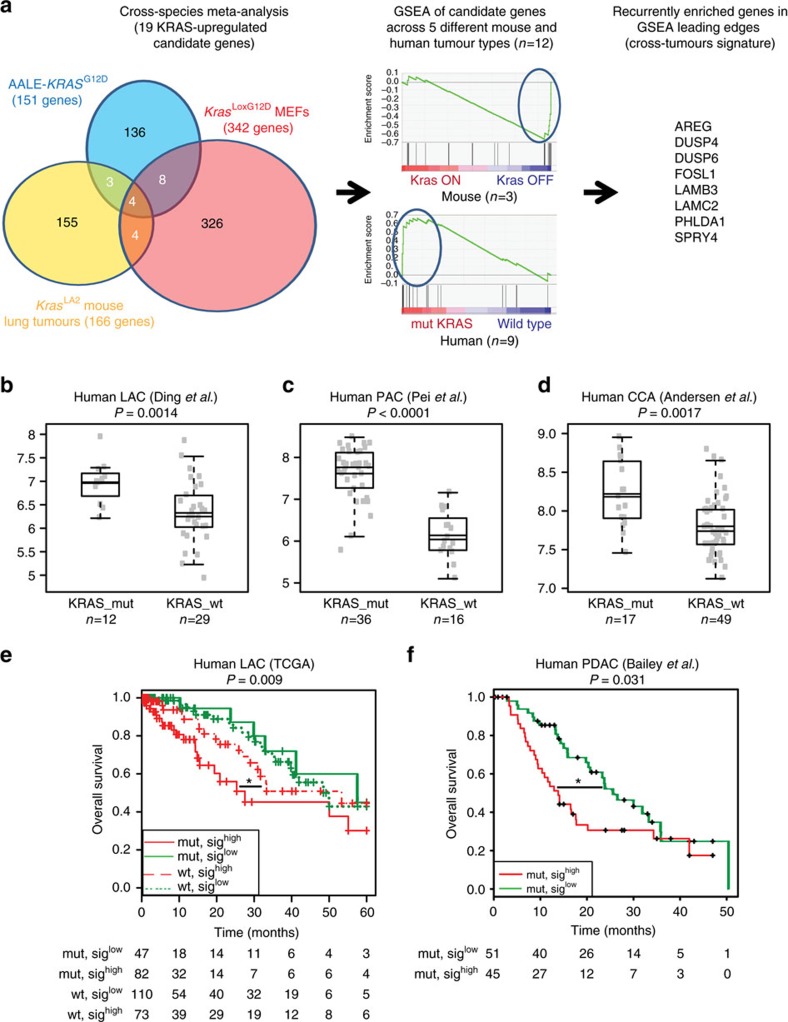
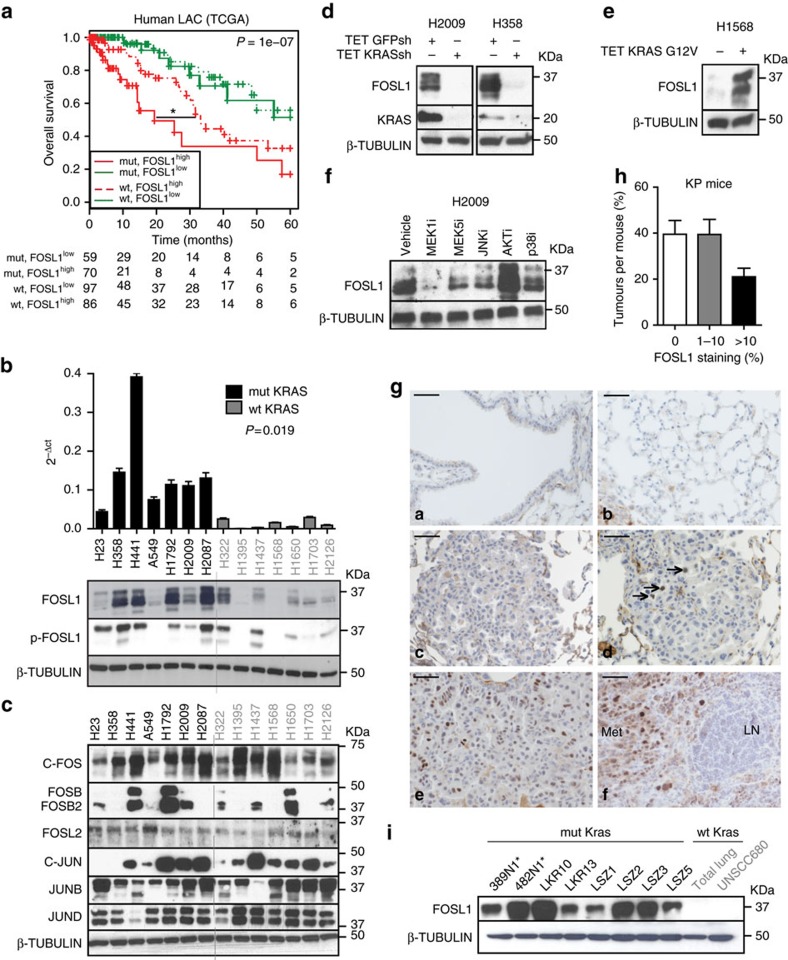
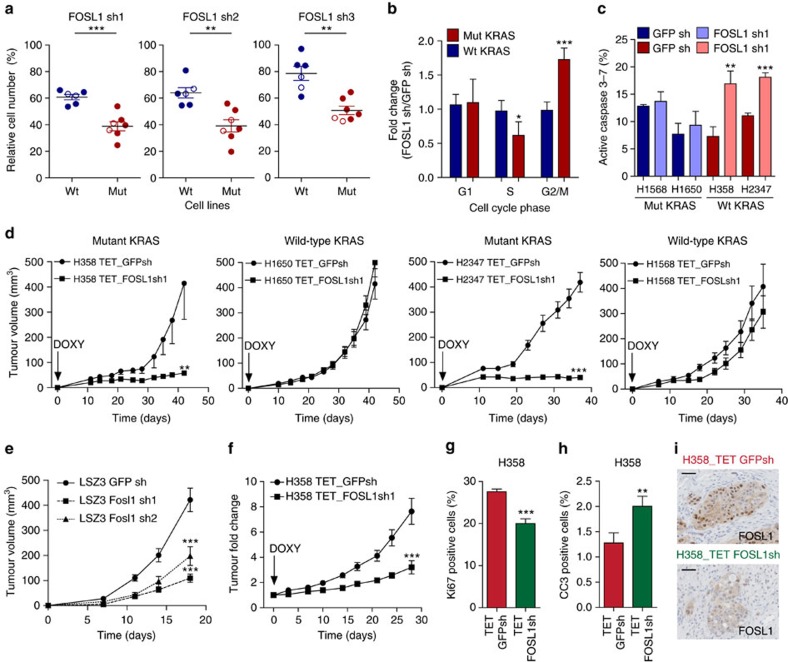
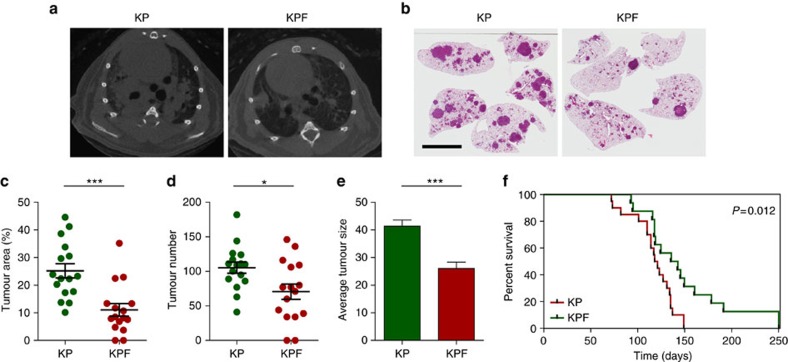
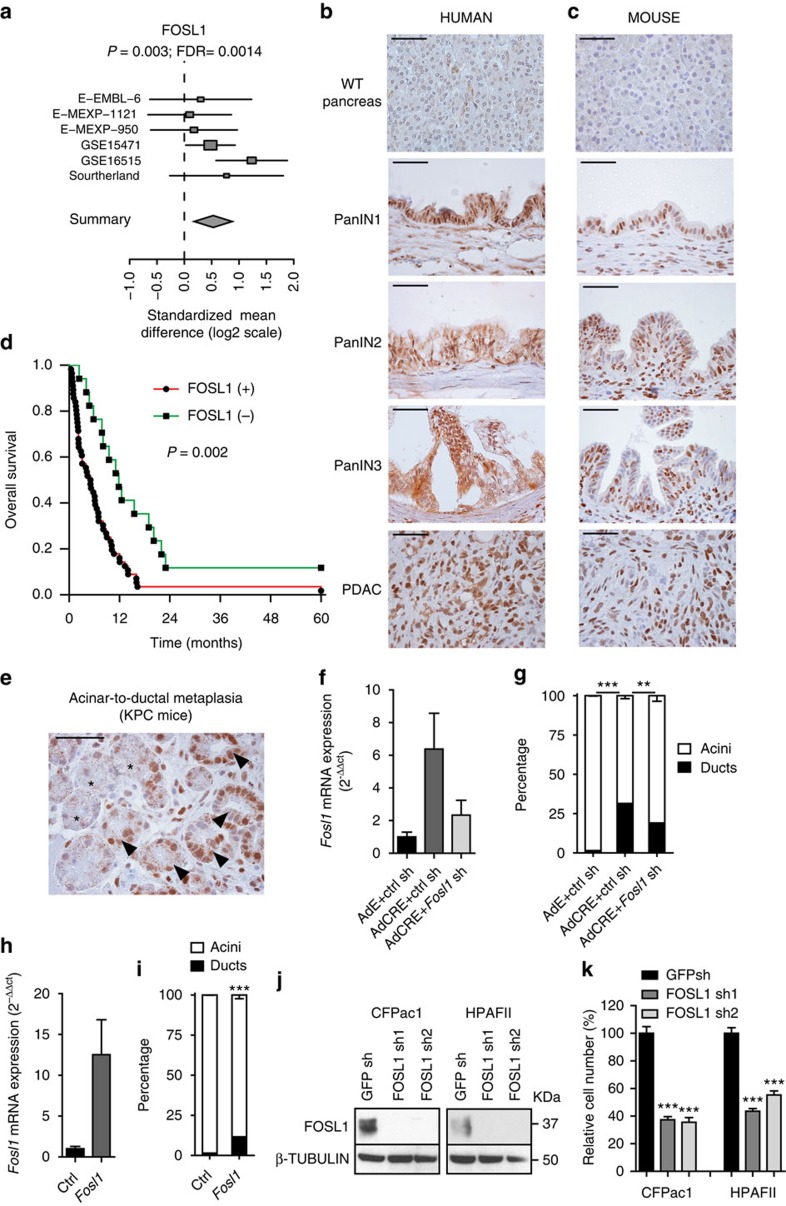
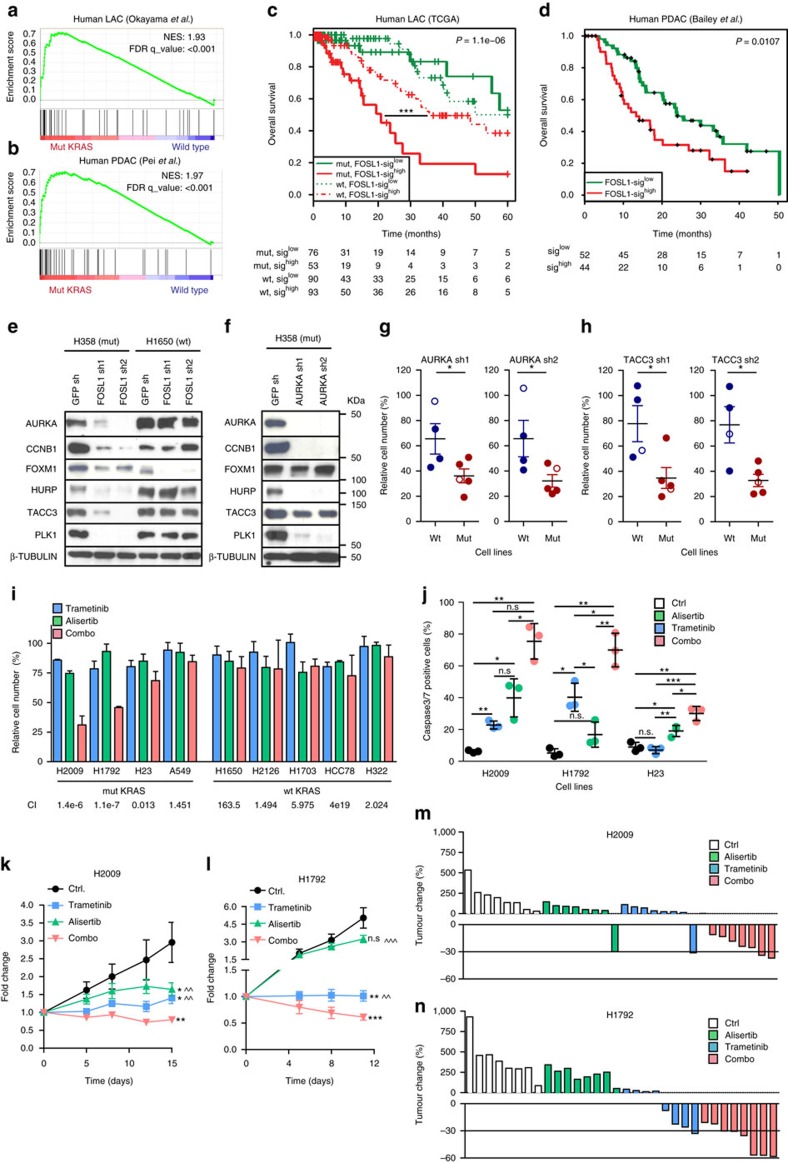
KRAS mutated tumours represent a large fraction of human cancers, but the vast majority remains refractory to current clinical therapies. Thus, a deeper understanding of the molecular mechanisms triggered by KRAS oncogene may yield alternative therapeutic strategies. Here we report the identification of a common transcriptional signature across mutant KRAS cancers of distinct tissue origin that includes the transcription factor FOSL1. High FOSL1 expression identifies mutant KRAS lung and pancreatic cancer patients with the worst survival outcome. Furthermore, FOSL1 genetic inhibition is detrimental to both KRAS-driven tumour types. Mechanistically, FOSL1 links the KRAS oncogene to components of the mitotic machinery, a pathway previously postulated to function orthogonally to oncogenic KRAS. FOSL1 targets include AURKA, whose inhibition impairs viability of mutant KRAS cells. Lastly, combination of AURKA and MEK inhibitors induces a deleterious effect on mutant KRAS cells. Our findings unveil KRAS downstream effectors that provide opportunities to treat KRAS-driven cancers.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
