

ALPK1 controls TIFA/TRAF6-dependent innate immunity against heptose-1,7-bisphosphate of gram-negative bacteria
- PMID: 28222186
- PMCID: PMC5336308
- DOI: 10.1371/journal.ppat.1006224
ALPK1 controls TIFA/TRAF6-dependent innate immunity against heptose-1,7-bisphosphate of gram-negative bacteria
Abstract
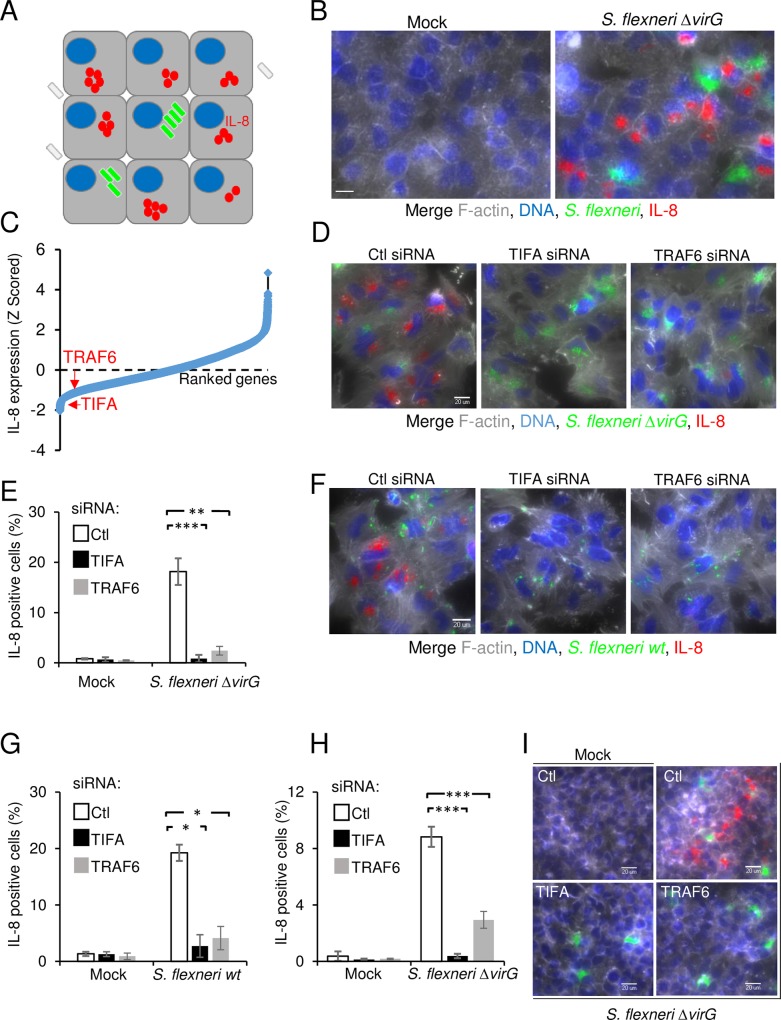
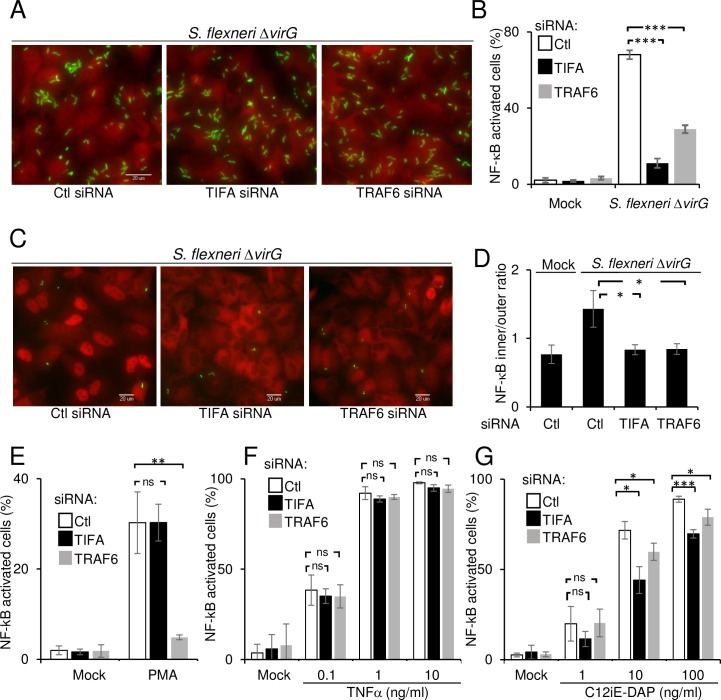
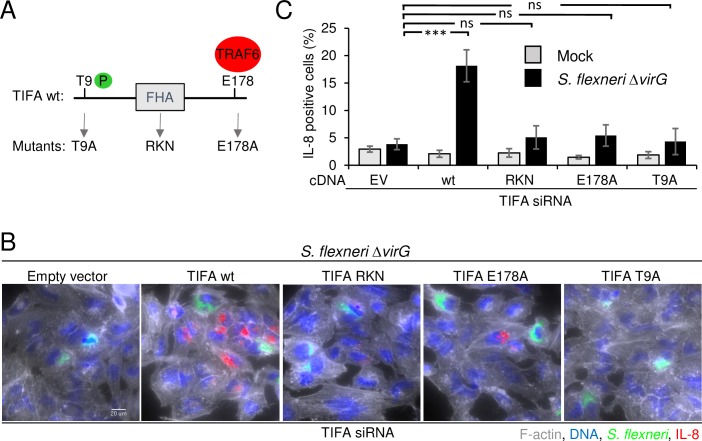
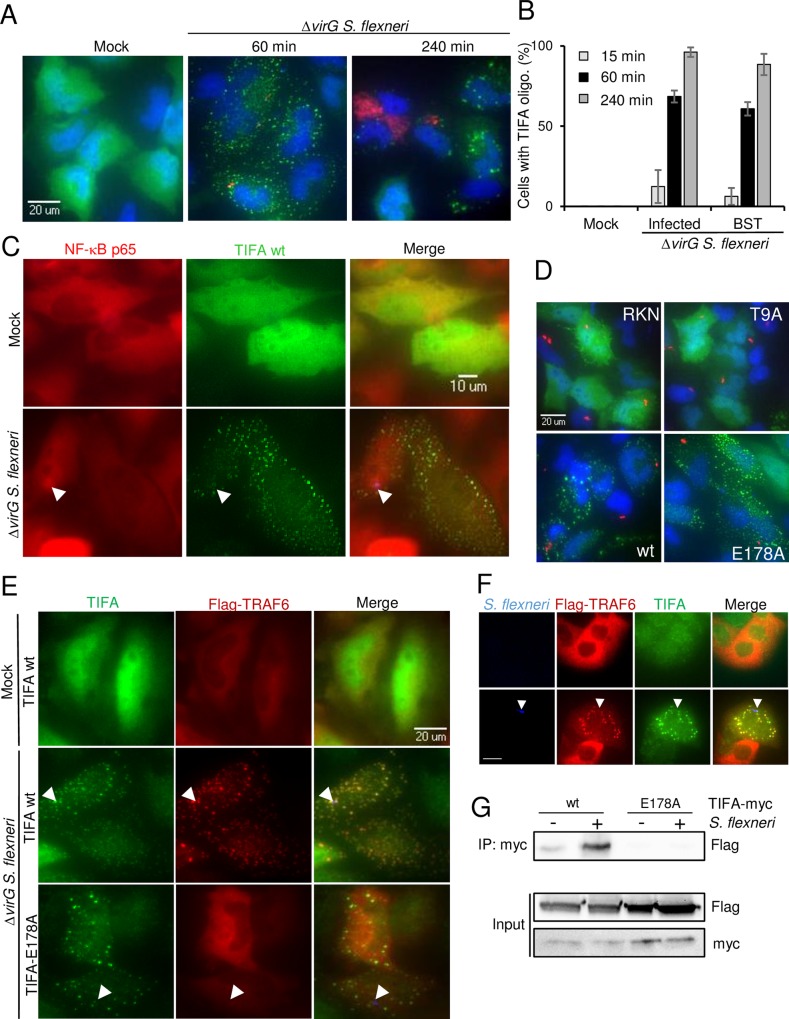
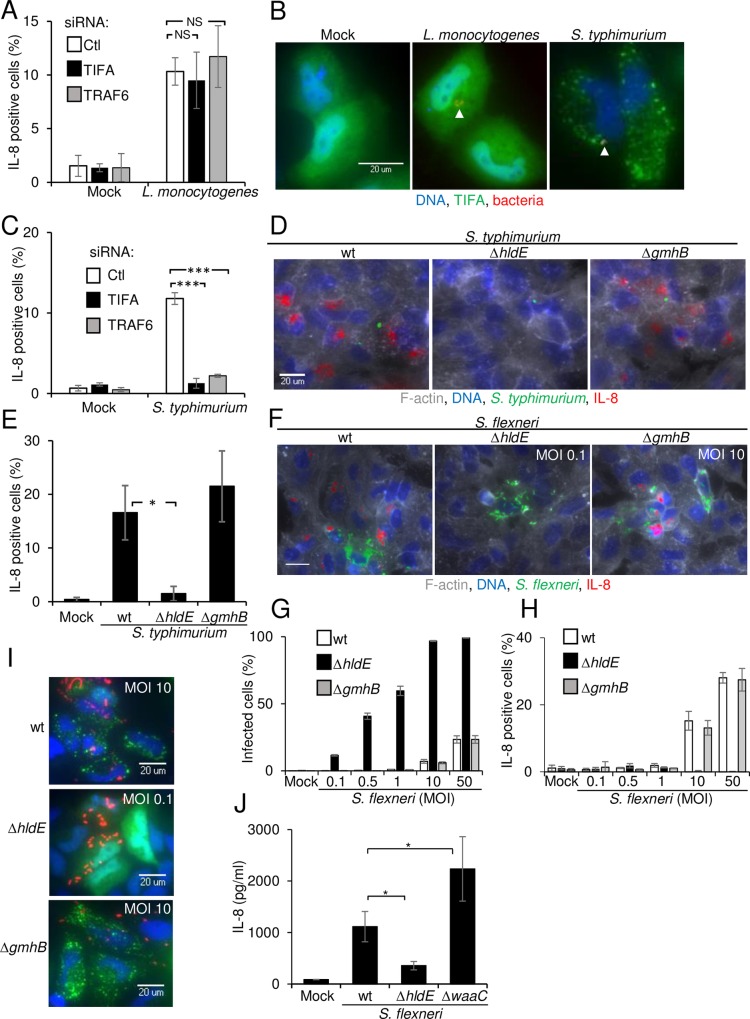
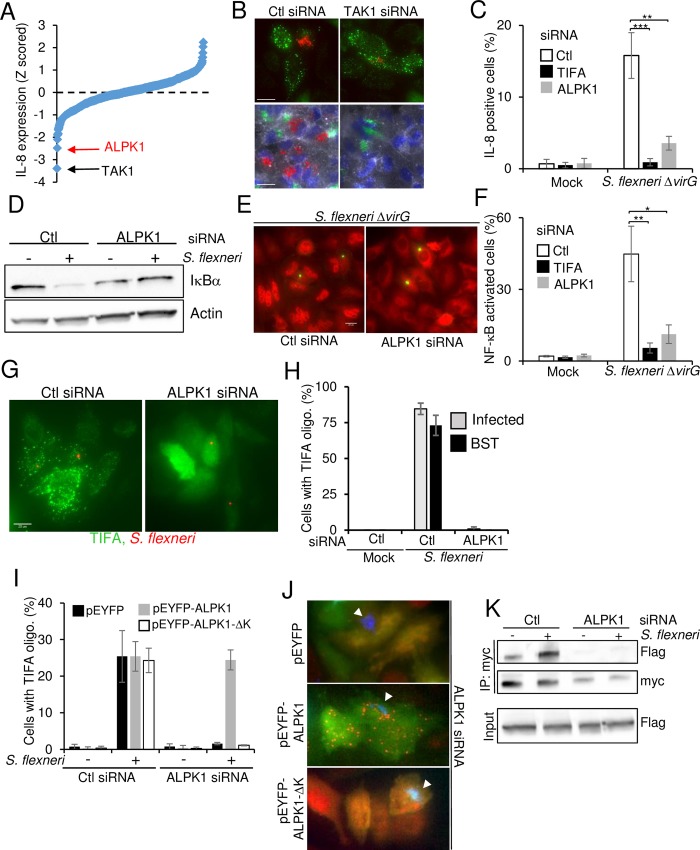
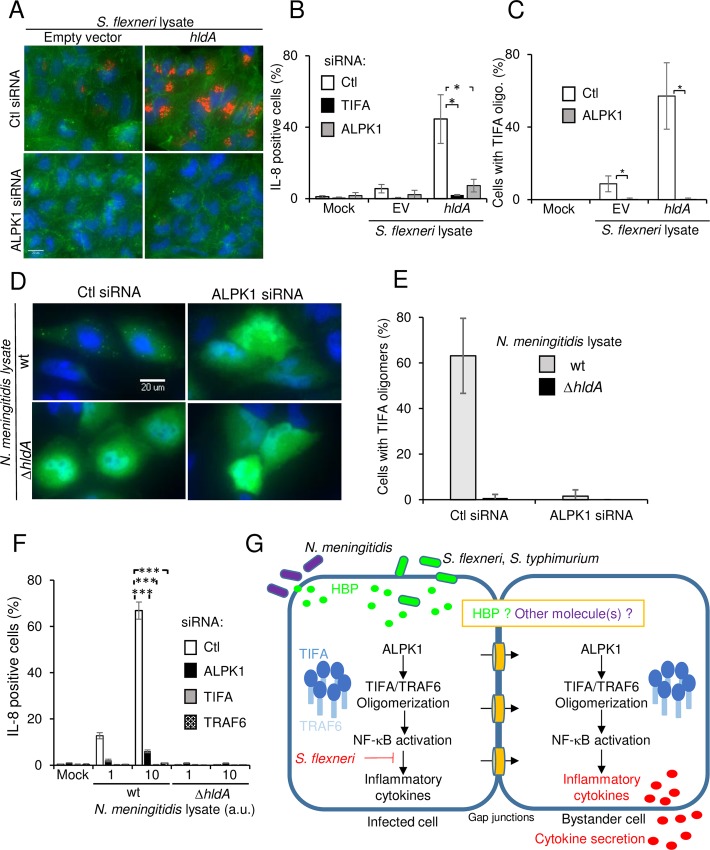
During infection by invasive bacteria, epithelial cells contribute to innate immunity via the local secretion of inflammatory cytokines. These are directly produced by infected cells or by uninfected bystanders via connexin-dependent cell-cell communication. However, the cellular pathways underlying this process remain largely unknown. Here we perform a genome-wide RNA interference screen and identify TIFA and TRAF6 as central players of Shigella flexneri and Salmonella typhimurium-induced interleukin-8 expression. We show that threonine 9 and the forkhead-associated domain of TIFA are necessary for the oligomerization of TIFA in both infected and bystander cells. Subsequently, this process triggers TRAF6 oligomerization and NF-κB activation. We demonstrate that TIFA/TRAF6-dependent cytokine expression is induced by the bacterial metabolite heptose-1,7-bisphosphate (HBP). In addition, we identify alpha-kinase 1 (ALPK1) as the critical kinase responsible for TIFA oligomerization and IL-8 expression in response to infection with S. flexneri and S. typhimurium but also to Neisseria meningitidis. Altogether, these results clearly show that ALPK1 is a master regulator of innate immunity against both invasive and extracellular gram-negative bacteria.
Conflict of interest statement
The authors have declared that no competing interests exist."
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
