

Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction
- PMID: 28228285
- PMCID: PMC5405381
- DOI: 10.1016/j.immuni.2017.02.007
Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction
Abstract
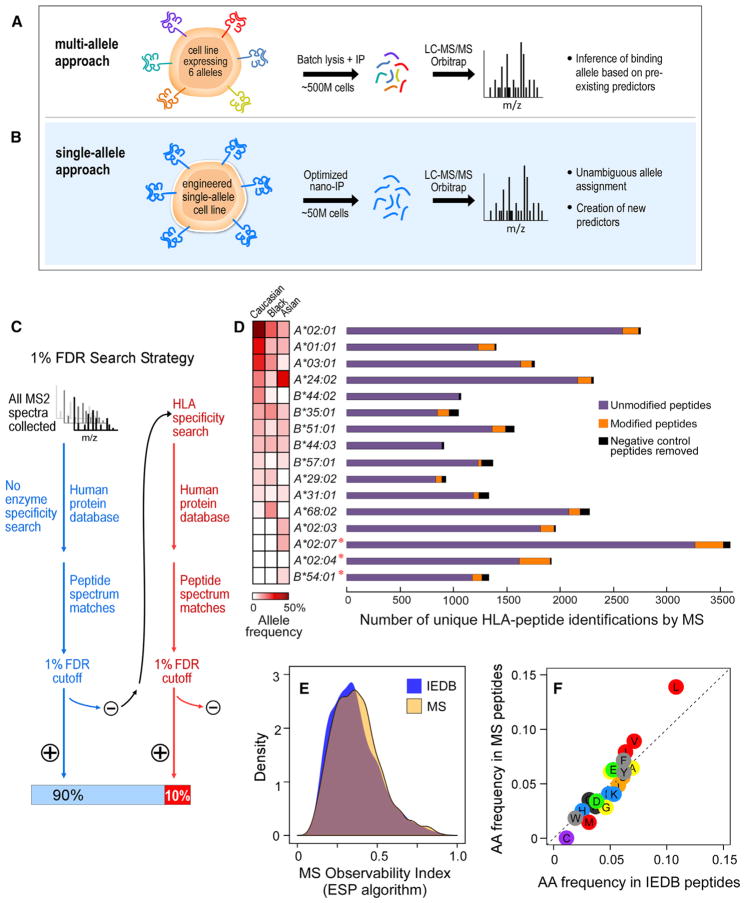
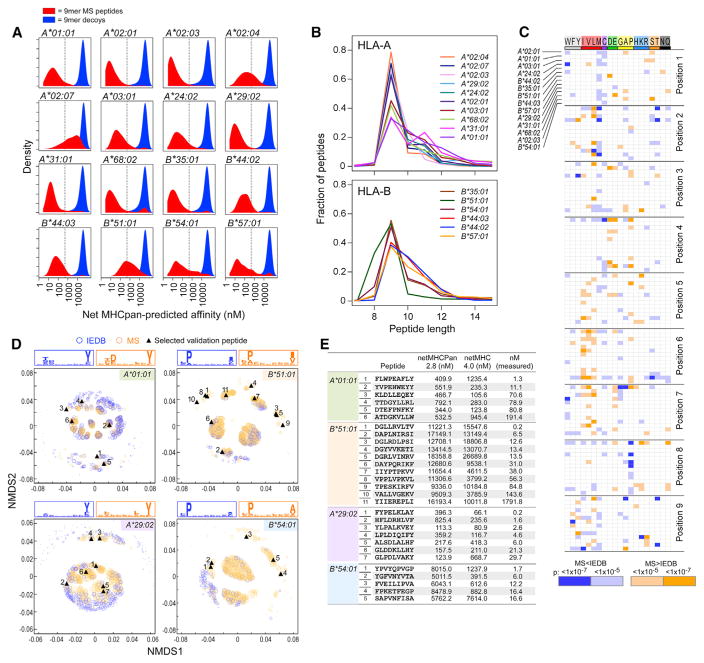
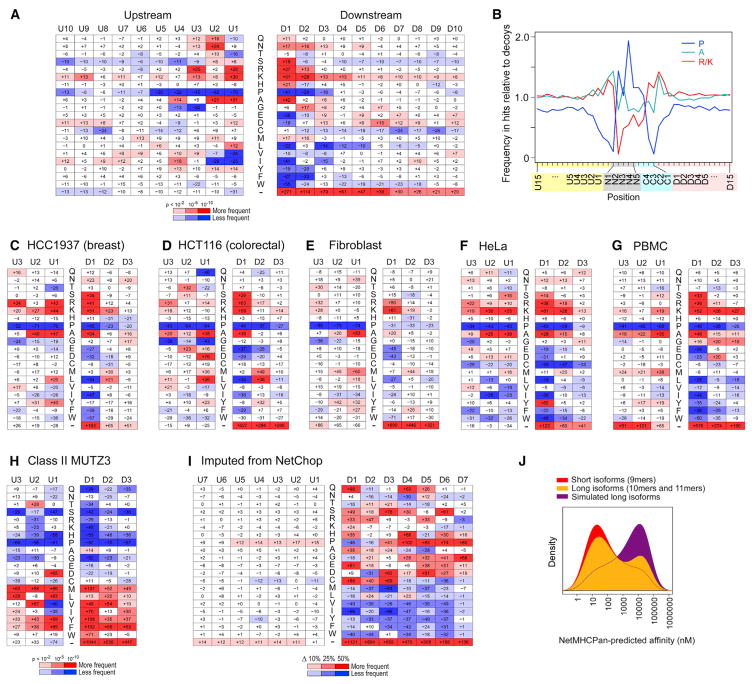
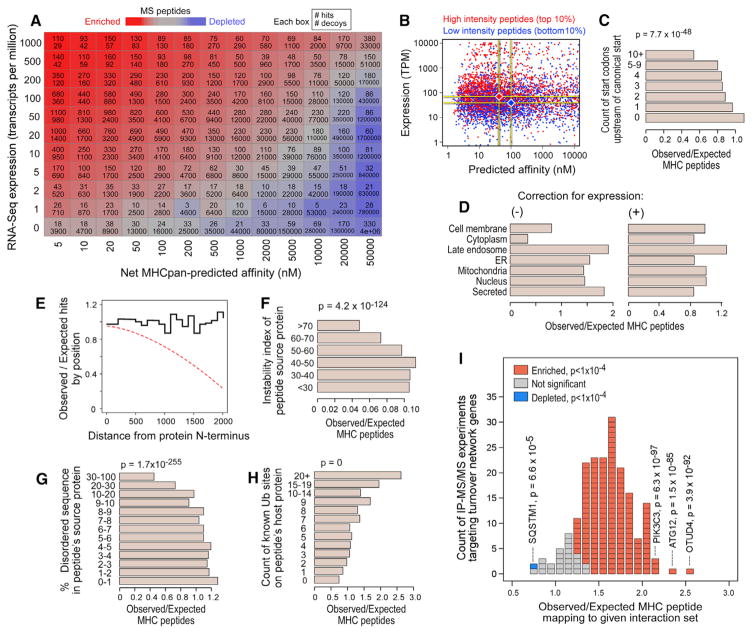
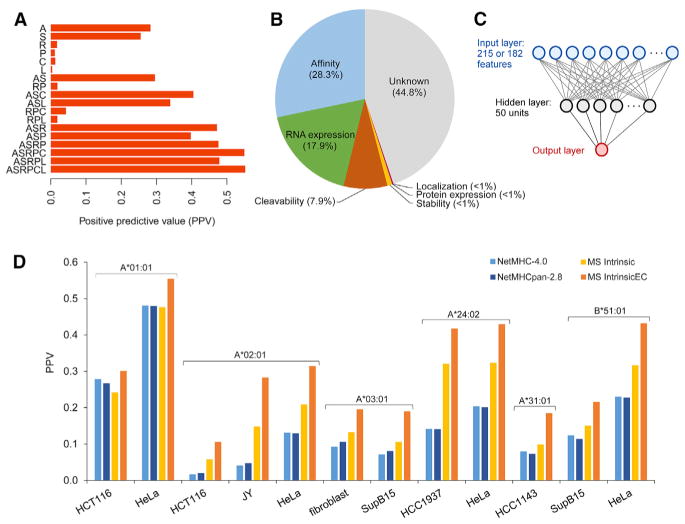
Identification of human leukocyte antigen (HLA)-bound peptides by liquid chromatography-tandem mass spectrometry (LC-MS/MS) is poised to provide a deep understanding of rules underlying antigen presentation. However, a key obstacle is the ambiguity that arises from the co-expression of multiple HLA alleles. Here, we have implemented a scalable mono-allelic strategy for profiling the HLA peptidome. By using cell lines expressing a single HLA allele, optimizing immunopurifications, and developing an application-specific spectral search algorithm, we identified thousands of peptides bound to 16 different HLA class I alleles. These data enabled the discovery of subdominant binding motifs and an integrative analysis quantifying the contribution of factors critical to epitope presentation, such as protein cleavage and gene expression. We trained neural-network prediction algorithms with our large dataset (>24,000 peptides) and outperformed algorithms trained on datasets of peptides with measured affinities. We thus demonstrate a strategy for systematically learning the rules of endogenous antigen presentation.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Bassani-Sternberg M, Gfeller D. Unsupervised HLA peptidome deconvolution improves ligand prediction accuracy and predicts cooperative effects in peptide-HLA interactions. J Immunol. 2016;197:2492–2499. - PubMed
-
- Berg M, Parbel A, Pettersen H, Fenyö D, Björkesten L. Detection of artifacts and peptide modifications in liquid chromatography/ mass spectrometry data using two-dimensional signal intensity map data visualization. Rapid Commun Mass Spectrom. 2006;20:1558–1562. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
