

Considerations and complications of mapping small RNA high-throughput data to transposable elements
- PMID: 28228849
- PMCID: PMC5311732
- DOI: 10.1186/s13100-017-0086-z
Considerations and complications of mapping small RNA high-throughput data to transposable elements
Abstract
Background: High-throughput sequencing (HTS) has revolutionized the way in which epigenetic research is conducted. When coupled with fully-sequenced genomes, millions of small RNA (sRNA) reads are mapped to regions of interest and the results scrutinized for clues about epigenetic mechanisms. However, this approach requires careful consideration in regards to experimental design, especially when one investigates repetitive parts of genomes such as transposable elements (TEs), or when such genomes are large, as is often the case in plants.
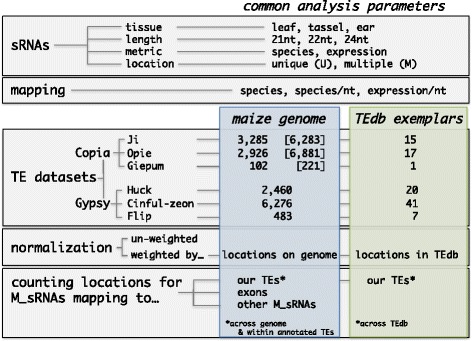
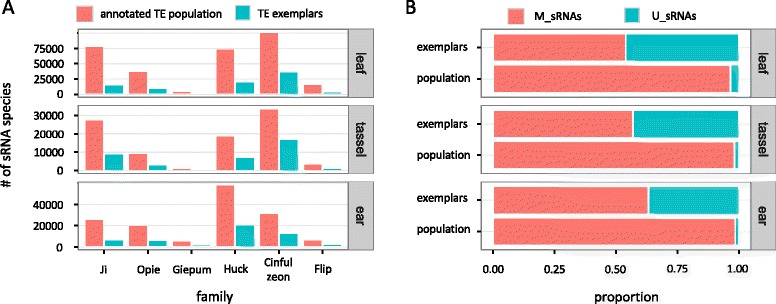
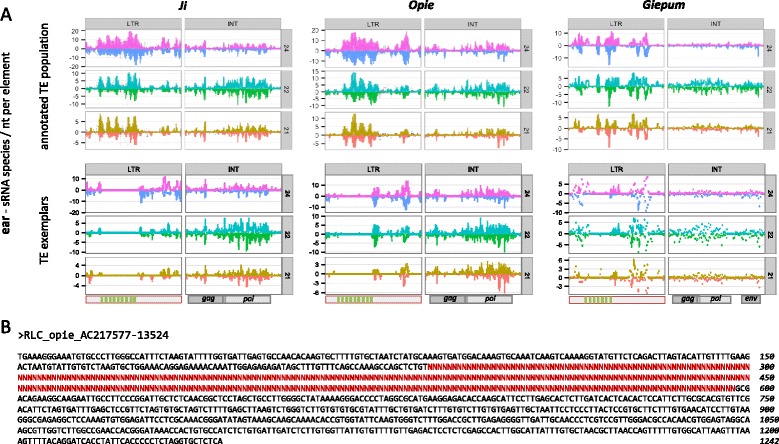
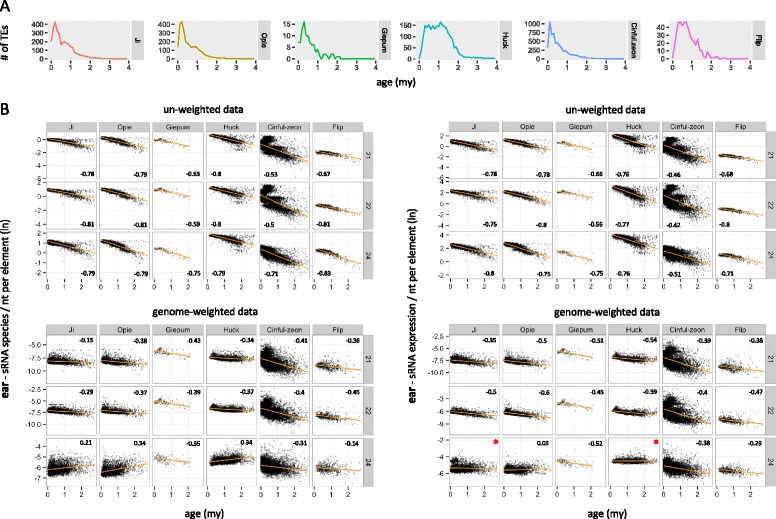
Results: Here, in an attempt to shed light on complications of mapping sRNAs to TEs, we focus on the 2,300 Mb maize genome, 85% of which is derived from TEs, and scrutinize methodological strategies that are commonly employed in TE studies. These include choices for the reference dataset, the normalization of multiply mapping sRNAs, and the selection among sRNA metrics. We further examine how these choices influence the relationship between sRNAs and the critical feature of TE age, and contrast their effect on low copy genomic regions and other popular HTS data.
Conclusions: Based on our analyses, we share a series of take-home messages that may help with the design, implementation, and interpretation of high-throughput TE epigenetic studies specifically, but our conclusions may also apply to any work that involves analysis of HTS data.
Keywords: Annotation; Bioinformatics; Genome mapping; High-throughput sequencing; RNA-seq; Small RNAs; Transposable elements; siRNAs.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
