

Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome
- PMID: 28229513
- PMCID: PMC5487276
- DOI: 10.1002/humu.23203
Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome
Abstract
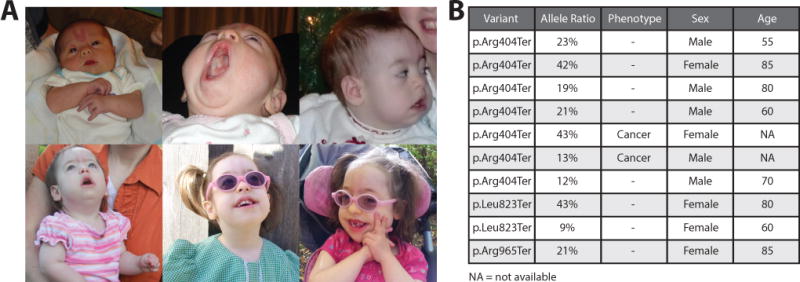
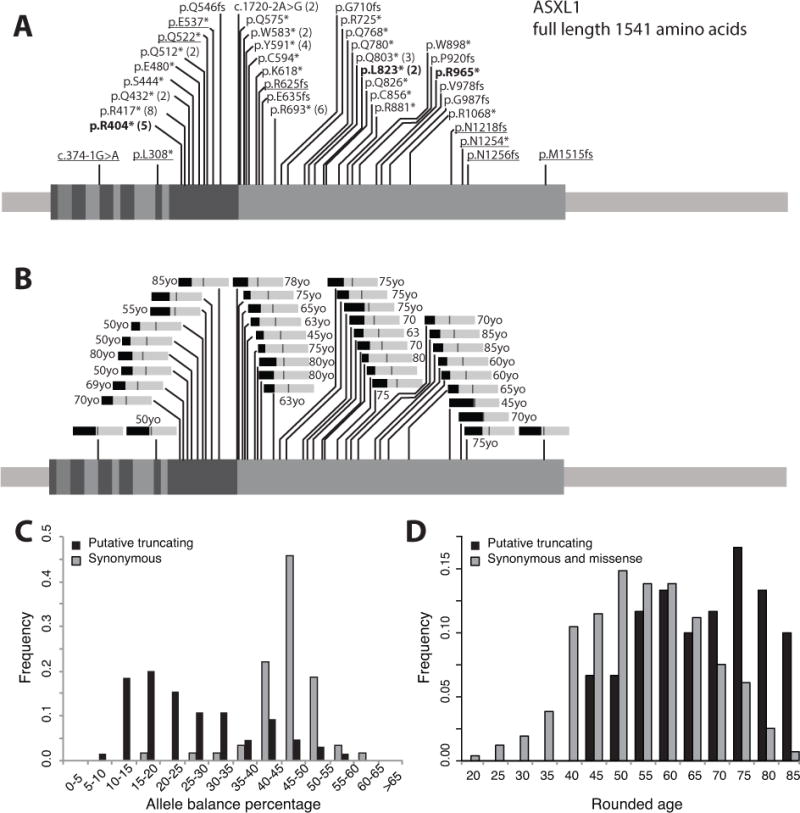
The clinical interpretation of genetic variants has come to rely heavily on reference population databases such as the Exome Aggregation Consortium (ExAC) database. Pathogenic variants in genes associated with severe, pediatric-onset, highly penetrant, autosomal dominant conditions are assumed to be absent or rare in these databases. Exome sequencing of a 6-year-old female patient with seizures, developmental delay, dysmorphic features, and failure to thrive identified an ASXL1 variant previously reported as causative of Bohring-Opitz syndrome (BOS). Surprisingly, the variant was observed seven times in the ExAC database, presumably in individuals without BOS. Although the BOS phenotype fit, the presence of the variant in reference population databases introduced ambiguity in result interpretation. Review of the literature revealed that acquired somatic mosaicism of ASXL1 variants (including pathogenic variants) during hematopoietic clonal expansion can occur with aging in healthy individuals. We examined all ASXL1 truncating variants in the ExAC database and determined most are likely somatic. Failure to consider somatic mosaicism may lead to the inaccurate assumption that conditions like BOS have reduced penetrance, or the misclassification of potentially pathogenic variants.
Keywords: ASXL1; Bohring-Opitz syndrome; DNMT3A; Exome Aggregation Consortium; Tatton-Brown-Rahman syndrome; clonal hematopoiesis of indeterminate potential; somatic mosaicism; variant interpretation.
© 2017 Wiley Periodicals, Inc.
Figures
Comment in
-
Bad blood contaminating germline databases?Hum Mutat. 2017 May;38(5):469. doi: 10.1002/humu.23217. Hum Mutat. 2017. PMID: 28425196 No abstract available.
References
-
- Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, Kuscu C, Hricik T, Ndiaye-Lobry D, Lafave LM, Koche R, Shih AH, Guryanova OA, Kim E, Li S, Pandey S, Shin JY, Telis L, Liu J, Bhatt PK, Monette S, Zhao X, Mason CE, Park CY, Bernstein BE, Aifantis I, Levine RL. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med. 2013;210:2641–2659. - PMC - PubMed
-
- Chen R, Shi L, Hakenberg J, Naughton B, Sklar P, Zhang J, Zhou H, Tian L, Prakash O, Lemire M, Sleiman P, Cheng W-Y, Chen W, Shah H, Shen Y, Fromer M, Omberg L, Deardorff MA, Zackai E, Bobe JR, Levin E, Hudson TJ, Groop L, Wang J, Hakonarson H, Wojcicki A, Diaz GA, Edelmann L, Schadt EE, Friend SH. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol. 2016;34:531–538. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
