

The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport
- PMID: 28232525
- PMCID: PMC5338353
- DOI: 10.1126/science.aag1789
The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport
Abstract
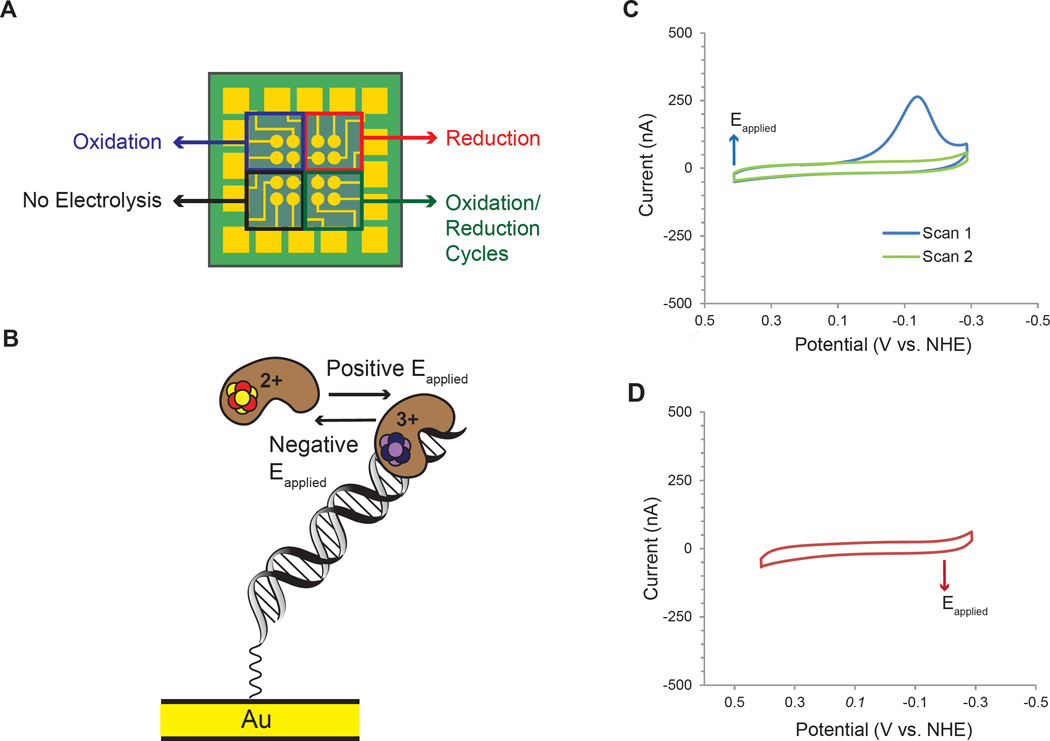
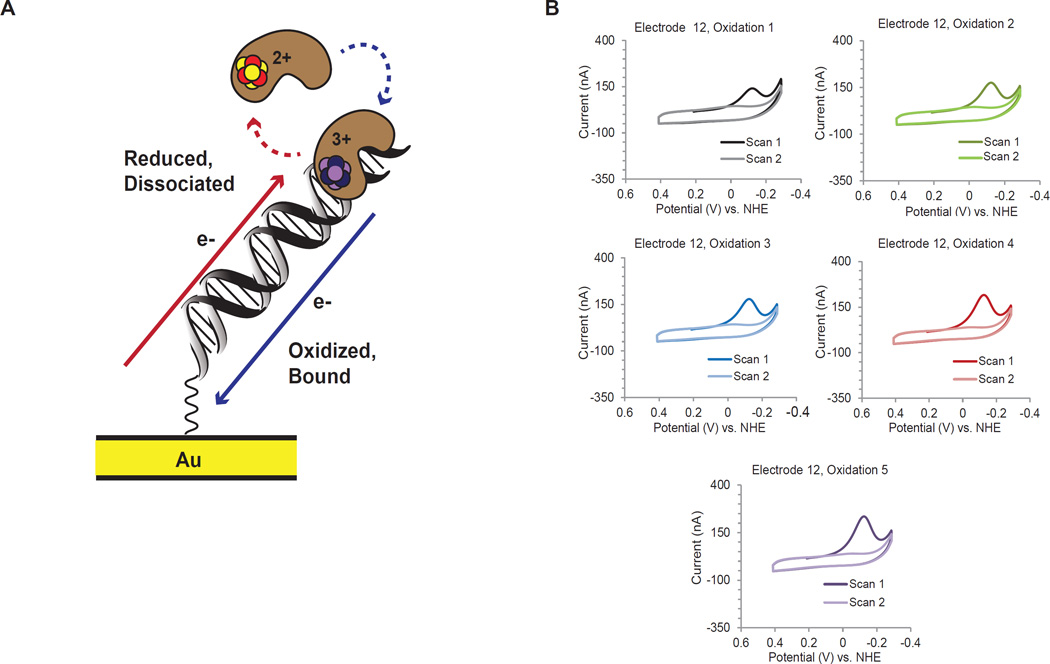
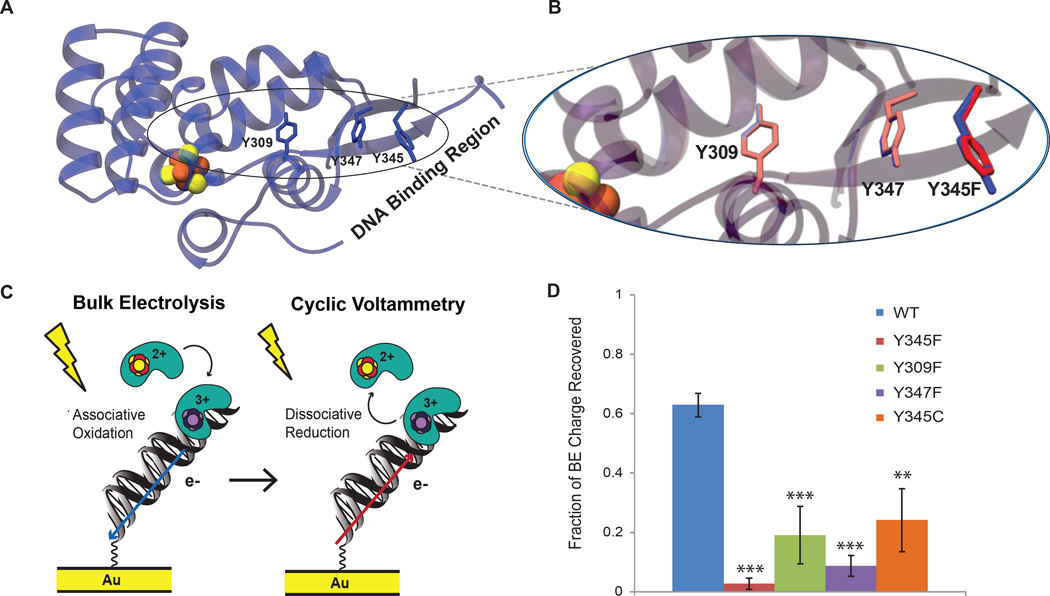
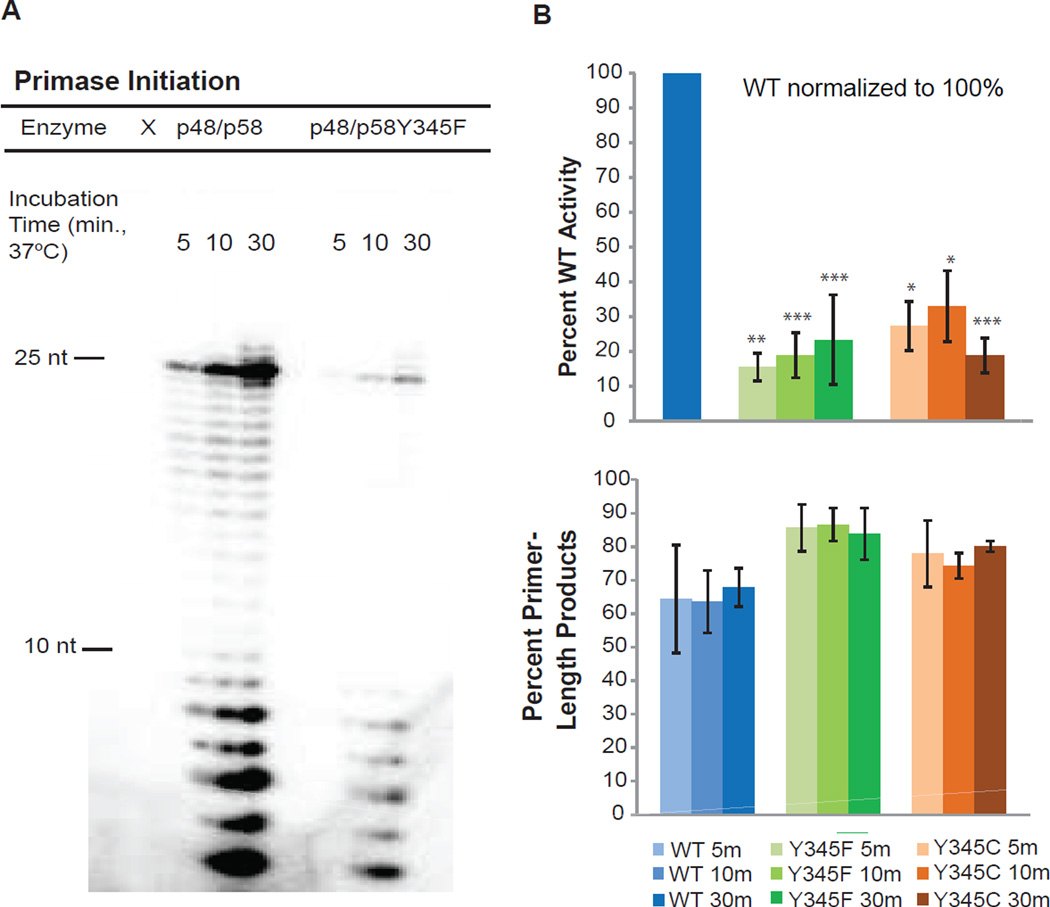
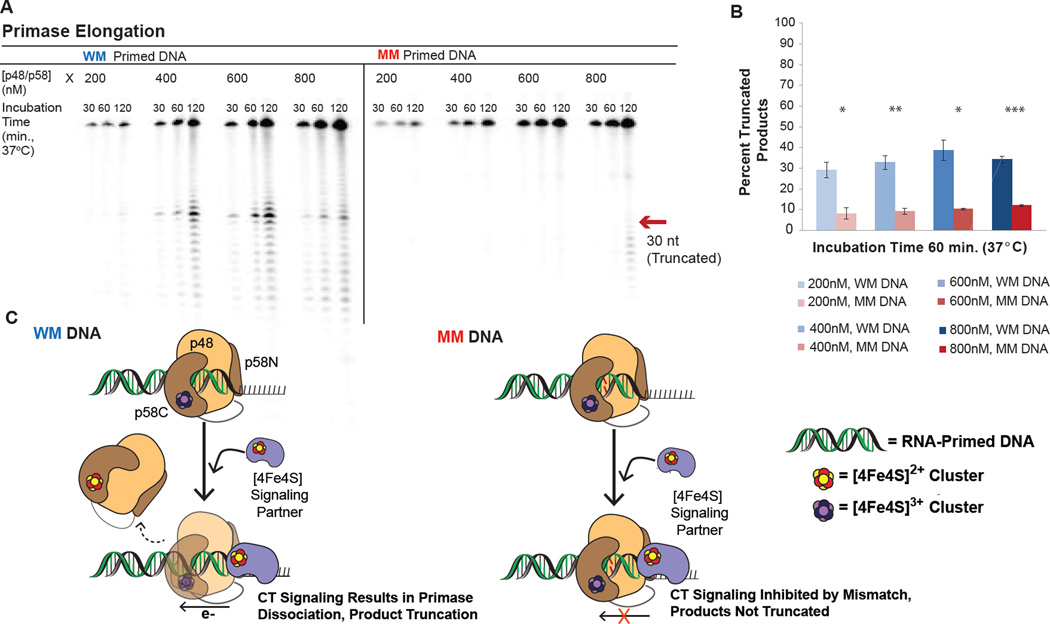
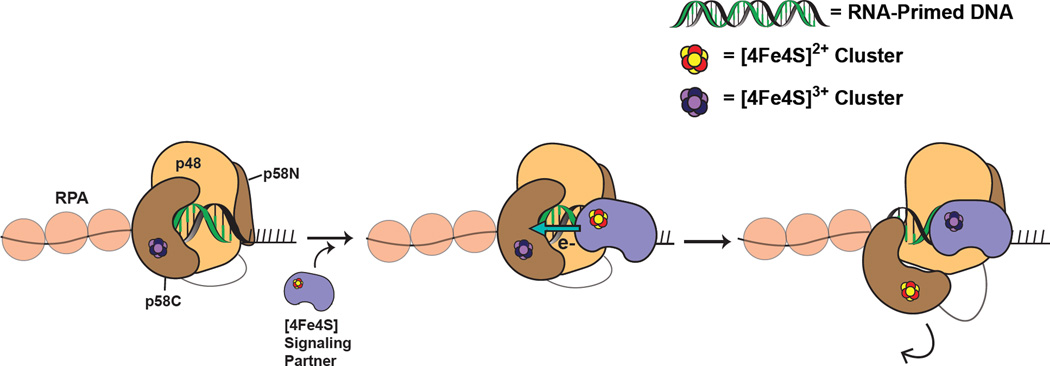
DNA charge transport chemistry offers a means of long-range, rapid redox signaling. We demonstrate that the [4Fe4S] cluster in human DNA primase can make use of this chemistry to coordinate the first steps of DNA synthesis. Using DNA electrochemistry, we found that a change in oxidation state of the [4Fe4S] cluster acts as a switch for DNA binding. Single-atom mutations that inhibit this charge transfer hinder primase initiation without affecting primase structure or polymerization. Generating a single base mismatch in the growing primer duplex, which attenuates DNA charge transport, inhibits primer truncation. Thus, redox signaling by [4Fe4S] clusters using DNA charge transport regulates primase binding to DNA and illustrates chemistry that may efficiently drive substrate handoff between polymerases during DNA replication.
Copyright © 2017, American Association for the Advancement of Science.
Figures
Comment in
-
Comment on "The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport".Science. 2017 Jul 21;357(6348):eaan2396. doi: 10.1126/science.aan2396. Science. 2017. PMID: 28729484 Free PMC article.
-
Comment on "The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport".Science. 2017 Jul 21;357(6348):eaan2954. doi: 10.1126/science.aan2954. Science. 2017. PMID: 28729486
References
-
- Gorodetsky AA, Boal AK, Barton JK. Direct electrochemistry of endonuclease III in the presence and absence of DNA. J. Am. Chem. Soc. 2006;128:12082–12083. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
