

CLK-dependent exon recognition and conjoined gene formation revealed with a novel small molecule inhibitor
- PMID: 28232751
- PMCID: PMC5431906
- DOI: 10.1038/s41467-016-0008-7
CLK-dependent exon recognition and conjoined gene formation revealed with a novel small molecule inhibitor
Abstract
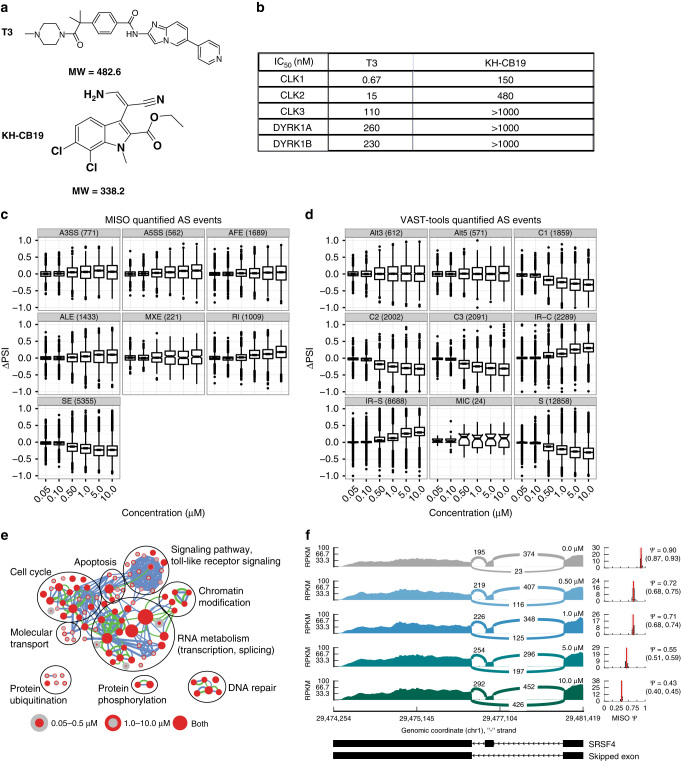
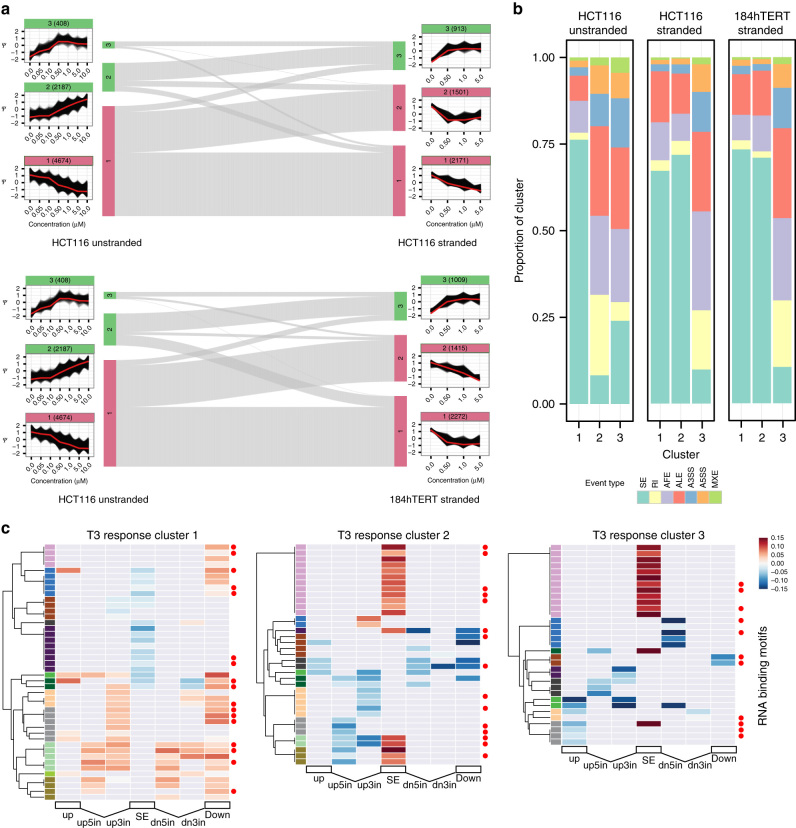
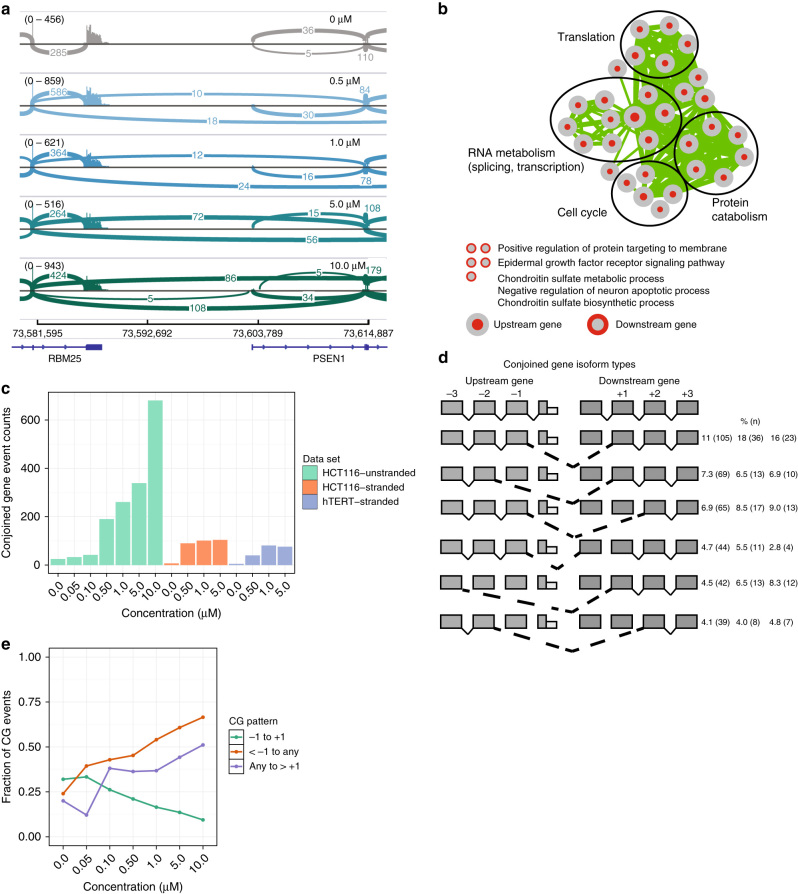
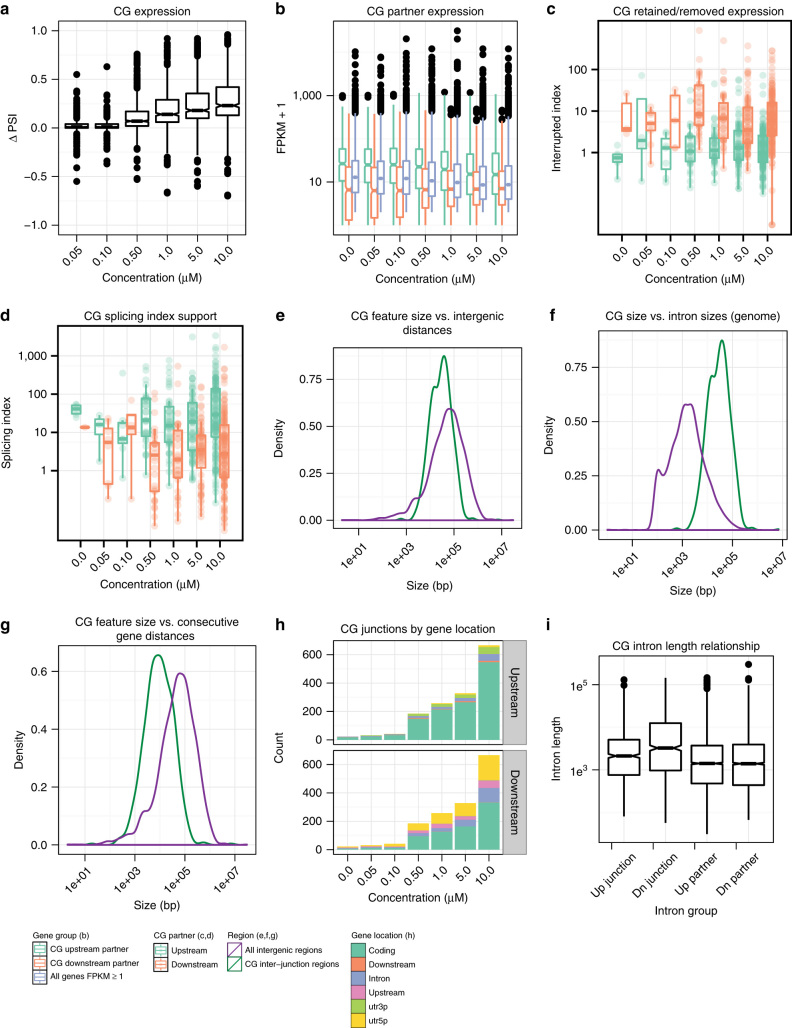
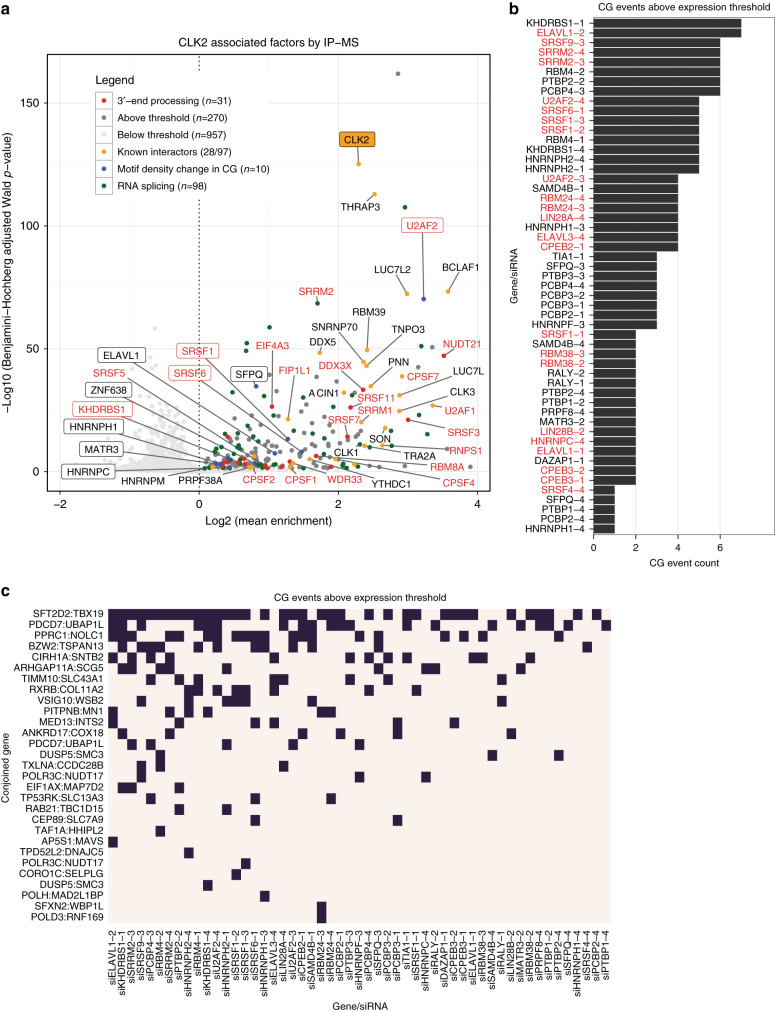
CDC-like kinase phosphorylation of serine/arginine-rich proteins is central to RNA splicing reactions. Yet, the genomic network of CDC-like kinase-dependent RNA processing events remains poorly defined. Here, we explore the connectivity of genomic CDC-like kinase splicing functions by applying graduated, short-exposure, pharmacological CDC-like kinase inhibition using a novel small molecule (T3) with very high potency, selectivity, and cell-based stability. Using RNA-Seq, we define CDC-like kinase-responsive alternative splicing events, the large majority of which monotonically increase or decrease with increasing CDC-like kinase inhibition. We show that distinct RNA-binding motifs are associated with T3 response in skipped exons. Unexpectedly, we observe dose-dependent conjoined gene transcription, which is associated with motif enrichment in the last and second exons of upstream and downstream partners, respectively. siRNA knockdown of CLK2-associated genes significantly increases conjoined gene formation. Collectively, our results reveal an unexpected role for CDC-like kinase in conjoined gene formation, via regulation of 3'-end processing and associated splicing factors.The phosphorylation of serine/arginine-rich proteins by CDC-like kinase is a central regulatory mechanism for RNA splicing reactions. Here, the authors synthesize a novel small molecule CLK inhibitor and map CLK-responsive alternative splicing events and discover an effect on conjoined gene transcription.
Figures

Similar articles
-
Akt2 regulation of Cdc2-like kinases (Clk/Sty), serine/arginine-rich (SR) protein phosphorylation, and insulin-induced alternative splicing of PKCbetaII messenger ribonucleic acid.Endocrinology. 2009 May;150(5):2087-97. doi: 10.1210/en.2008-0818. Epub 2008 Dec 30. Endocrinology. 2009. PMID: 19116344 Free PMC article.
-
Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability.EMBO Mol Med. 2018 Jun;10(6):e8289. doi: 10.15252/emmm.201708289. EMBO Mol Med. 2018. PMID: 29769258 Free PMC article.
-
Cdc2-like kinases and DNA topoisomerase I regulate alternative splicing of tissue factor in human endothelial cells.Circ Res. 2009 Mar 13;104(5):589-99. doi: 10.1161/CIRCRESAHA.108.183905. Epub 2009 Jan 22. Circ Res. 2009. PMID: 19168442
-
Human CDC2-like kinase 1 (CLK1): a novel target for Alzheimer's disease.Curr Drug Targets. 2014 May;15(5):539-50. doi: 10.2174/1389450115666140226112321. Curr Drug Targets. 2014. PMID: 24568585 Review.
-
Cdc-Like Kinases (CLKs): Biology, Chemical Probes, and Therapeutic Potential.Int J Mol Sci. 2020 Oct 13;21(20):7549. doi: 10.3390/ijms21207549. Int J Mol Sci. 2020. PMID: 33066143 Free PMC article. Review.
Cited by
-
Pharmacological systems analysis defines EIF4A3 functions in cell-cycle and RNA stress granule formation.Commun Biol. 2019 May 3;2:165. doi: 10.1038/s42003-019-0391-9. eCollection 2019. Commun Biol. 2019. PMID: 31069274 Free PMC article.
-
Targeting pre-mRNA splicing in cancers: roles, inhibitors, and therapeutic opportunities.Front Oncol. 2023 Jun 5;13:1152087. doi: 10.3389/fonc.2023.1152087. eCollection 2023. Front Oncol. 2023. PMID: 37342192 Free PMC article. Review.
-
Exon inclusion signatures enable accurate estimation of splicing factor activity.bioRxiv [Preprint]. 2025 Jan 30:2024.06.21.600051. doi: 10.1101/2024.06.21.600051. bioRxiv. 2025. PMID: 38979366 Free PMC article. Preprint.
-
Gene Fusion Detection and Characterization in Long-Read Cancer Transcriptome Sequencing Data with FusionSeeker.Cancer Res. 2023 Jan 4;83(1):28-33. doi: 10.1158/0008-5472.CAN-22-1628. Cancer Res. 2023. PMID: 36318117 Free PMC article.
-
Synergistic apoptotic effects in cancer cells by the combination of CLK and Bcl-2 family inhibitors.PLoS One. 2020 Oct 16;15(10):e0240718. doi: 10.1371/journal.pone.0240718. eCollection 2020. PLoS One. 2020. PMID: 33064779 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
