

Bloom's Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition
- PMID: 28232778
- PMCID: PMC5260600
- DOI: 10.1159/000452082
Bloom's Syndrome: Clinical Spectrum, Molecular Pathogenesis, and Cancer Predisposition
Abstract
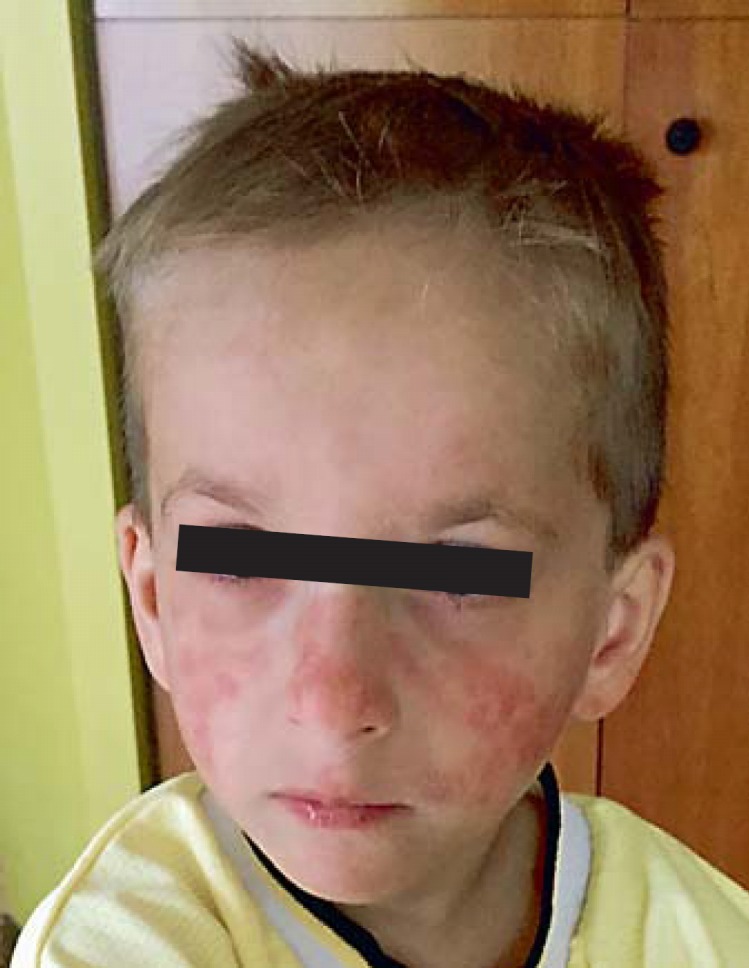
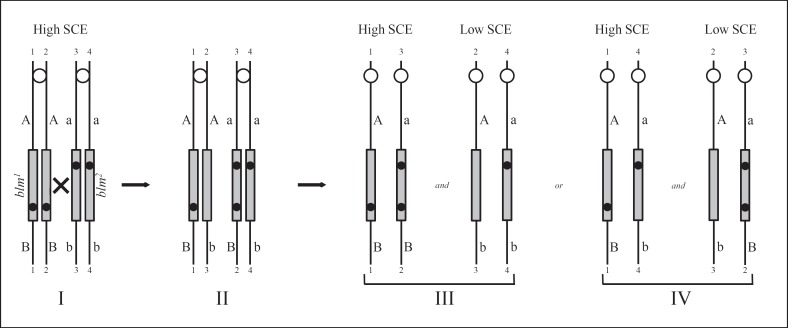
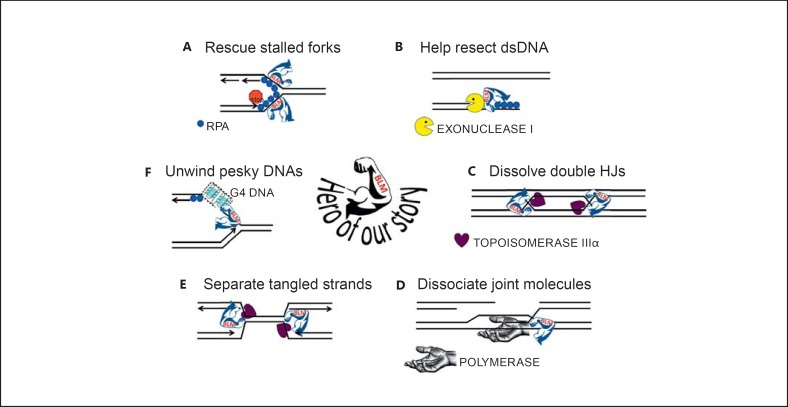
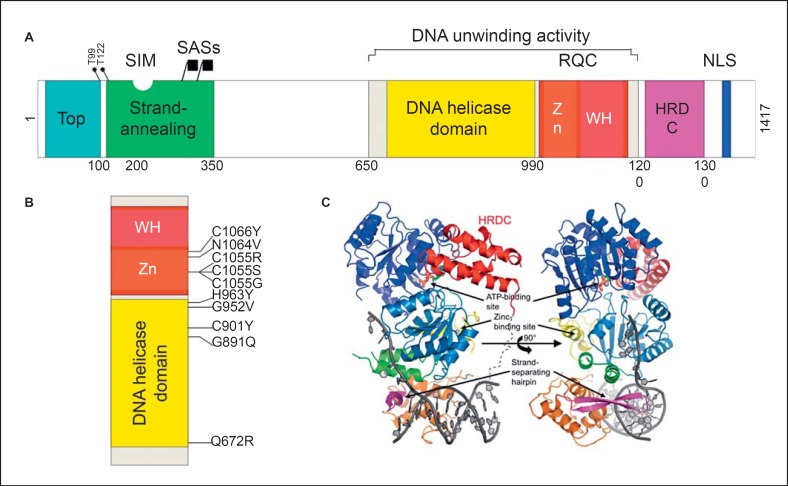
Bloom's syndrome is an autosomal recessive disorder characterized by prenatal and postnatal growth deficiency, photosensitive skin changes, immune deficiency, insulin resistance, and a greatly increased risk of early onset of cancer and for the development of multiple cancers. Loss-of-function mutations of BLM, which codes for a RecQ helicase, cause Bloom's syndrome. The absence of a functional BLM protein causes chromosome instability, excessive homologous recombination, and a greatly increased number of sister chromatid exchanges that are pathognomonic of the syndrome. A common founder mutation designated blmAsh is present in about 1 in 100 persons of Eastern European Jewish ancestry, and there are additional recurrent founder mutations among other populations. Missense, nonsense, and frameshift mutations as well as multiexonic deletions have all been observed. Bloom's syndrome is a prototypical chromosomal instability syndrome, and the somatic mutations that occur as a result of that instability are responsible for the increased cancer risk. Although there is currently no treatment aimed at the underlying genetic abnormality, persons with Bloom's syndrome benefit from sun protection, aggressive treatment of infections, surveillance for insulin resistance, and early identification of cancer.
Keywords: BLM; Bloom's syndrome; Cancer.
Figures
References
-
- Adams MD, McVey M, Sekelsky JJ. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science. 2003;299:265–267. - PubMed
-
- Antczak A, Kluźniak A, Kashyap A, Jakubowska A, et al. A common nonsense mutation of the BLM gene and prostate cancer risk and survival. Gene. 2013;532:173–1766. - PubMed
-
- Arora H, Chacon AH, Choudhary S, McLeod MP, Meshkov L, et al. Bloom syndrome. Int J Dermatol. 2014;53:798–802. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
