

SCN3A deficiency associated with increased seizure susceptibility
- PMID: 28235671
- PMCID: PMC5446790
- DOI: 10.1016/j.nbd.2017.02.006
SCN3A deficiency associated with increased seizure susceptibility
Abstract
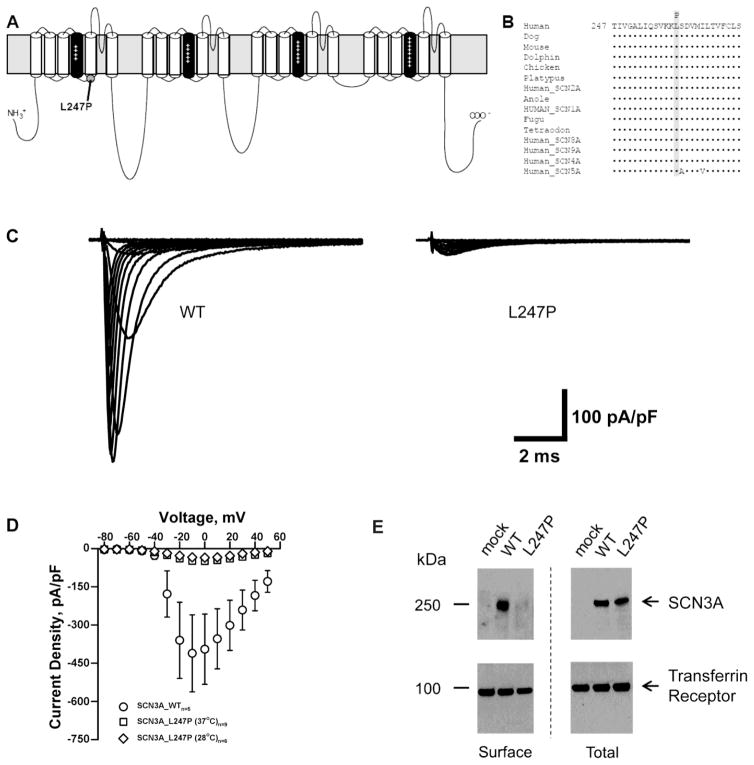
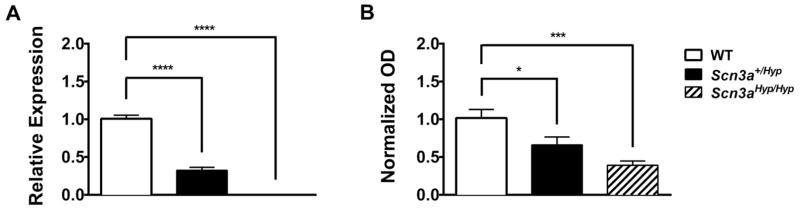
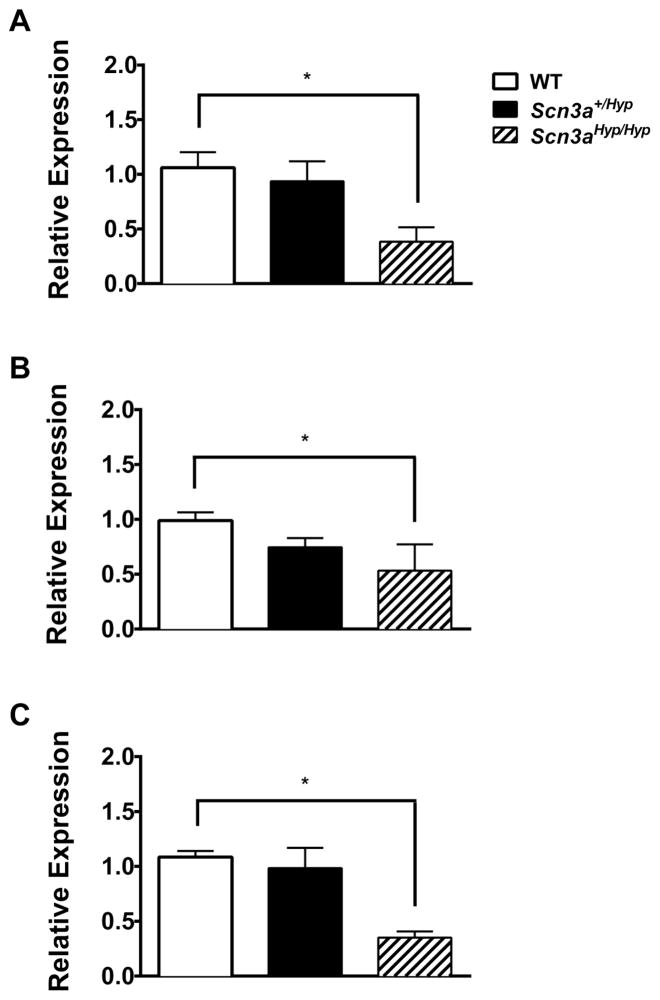
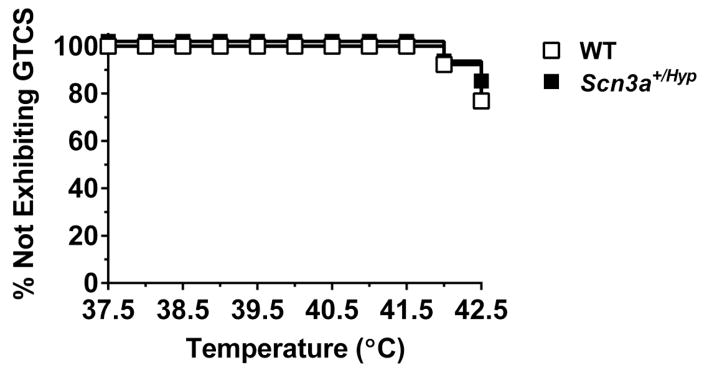
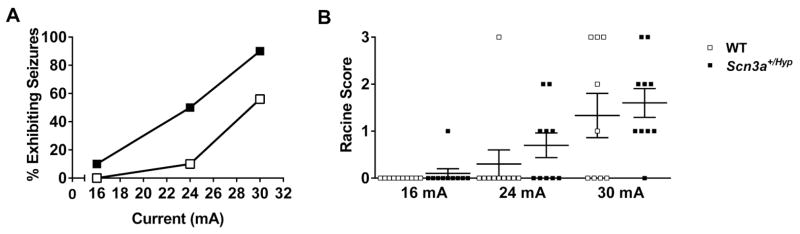
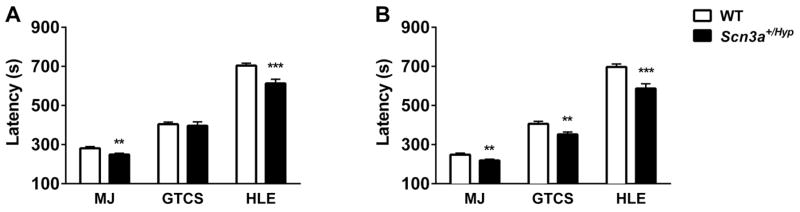
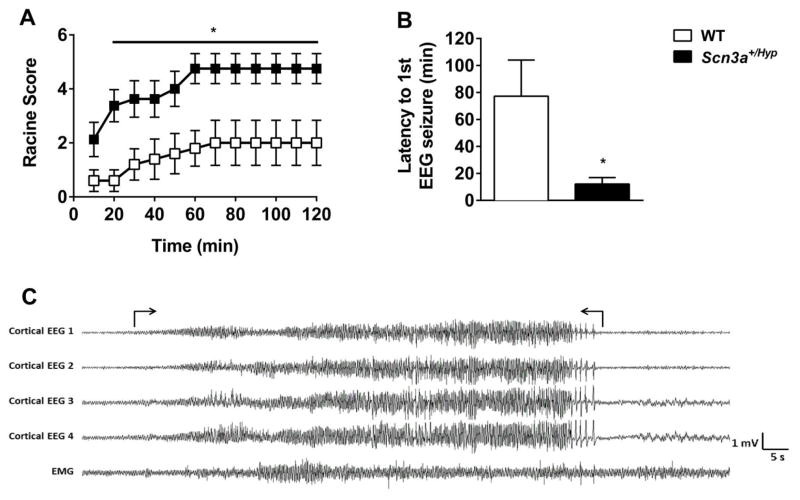
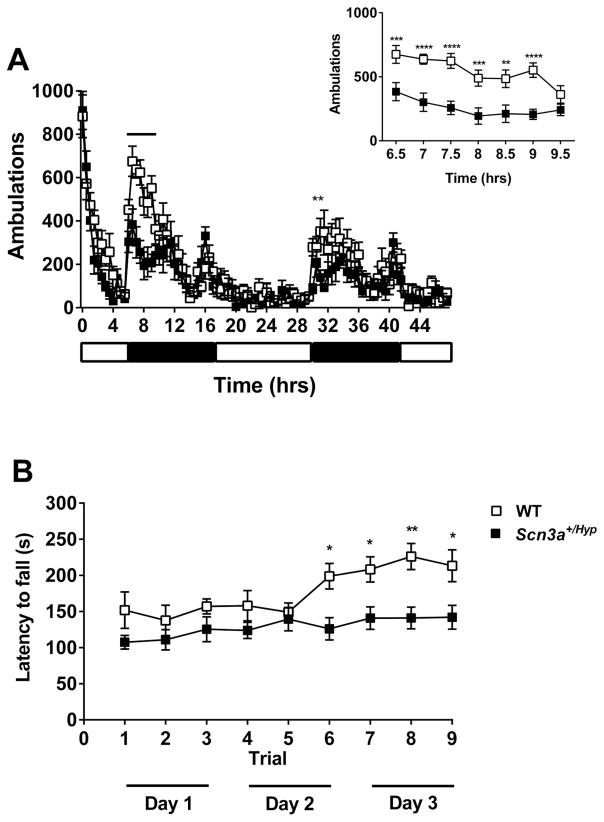
Mutations in voltage-gated sodium channels expressed highly in the brain (SCN1A, SCN2A, SCN3A, and SCN8A) are responsible for an increasing number of epilepsy syndromes. In particular, mutations in the SCN3A gene, encoding the pore-forming Nav1.3 α subunit, have been identified in patients with focal epilepsy. Biophysical characterization of epilepsy-associated SCN3A variants suggests that both gain- and loss-of-function SCN3A mutations may lead to increased seizure susceptibility. In this report, we identified a novel SCN3A variant (L247P) by whole exome sequencing of a child with focal epilepsy, developmental delay, and autonomic nervous system dysfunction. Voltage clamp analysis showed no detectable sodium current in a heterologous expression system expressing the SCN3A-L247P variant. Furthermore, cell surface biotinylation demonstrated a reduction in the amount of SCN3A-L247P at the cell surface, suggesting the SCN3A-L247P variant is a trafficking-deficient mutant. To further explore the possible clinical consequences of reduced SCN3A activity, we investigated the effect of a hypomorphic Scn3a allele (Scn3aHyp) on seizure susceptibility and behavior using a gene trap mouse line. Heterozygous Scn3a mutant mice (Scn3a+/Hyp) did not exhibit spontaneous seizures nor were they susceptible to hyperthermia-induced seizures. However, they displayed increased susceptibility to electroconvulsive (6Hz) and chemiconvulsive (flurothyl and kainic acid) induced seizures. Scn3a+/Hyp mice also exhibited deficits in locomotor activity and motor learning. Taken together, these results provide evidence that loss-of-function of SCN3A caused by reduced protein expression or deficient trafficking to the plasma membrane may contribute to increased seizure susceptibility.
Keywords: Focal epilepsy; Na(v)1.3; SCN3A; Seizure susceptibility; Voltage-gated sodium channel.
Copyright © 2017 Elsevier Inc. All rights reserved.
Figures
References
-
- Baasch AL, Huning I, Gilissen C, Klepper J, Veltman JA, Gillessen-Kaesbach G, Hoischen A, Lohmann K. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia. 2014;55:e25–9. - PubMed
-
- Barton ME, Klein BD, Wolf HH, White HS. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001;47:217–27. - PubMed
-
- Bechi G, Rusconi R, Cestele S, Striano P, Franceschetti S, Mantegazza M. Rescuable folding defective NaV1.1 (SCN1A) mutants in epilepsy: Properties, occurrence, and novel rescuing strategy with peptides targeted to the endoplasmic reticulum. Neurobiol Dis. 2015;75:100–14. - PubMed
-
- Black JA, Yokoyama S, Higashida H, Ransom BR, Waxman SG. Sodium channel mRNAs I, II and III in the CNS: cell-specific expression. Brain Res Mol Brain Res. 1994;22:275–89. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- R01 NS090319/NS/NINDS NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- R01 NS072221/NS/NINDS NIH HHS/United States
- P30 CA060553/CA/NCI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- R01 NS053792/NS/NINDS NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
